The Tools menu
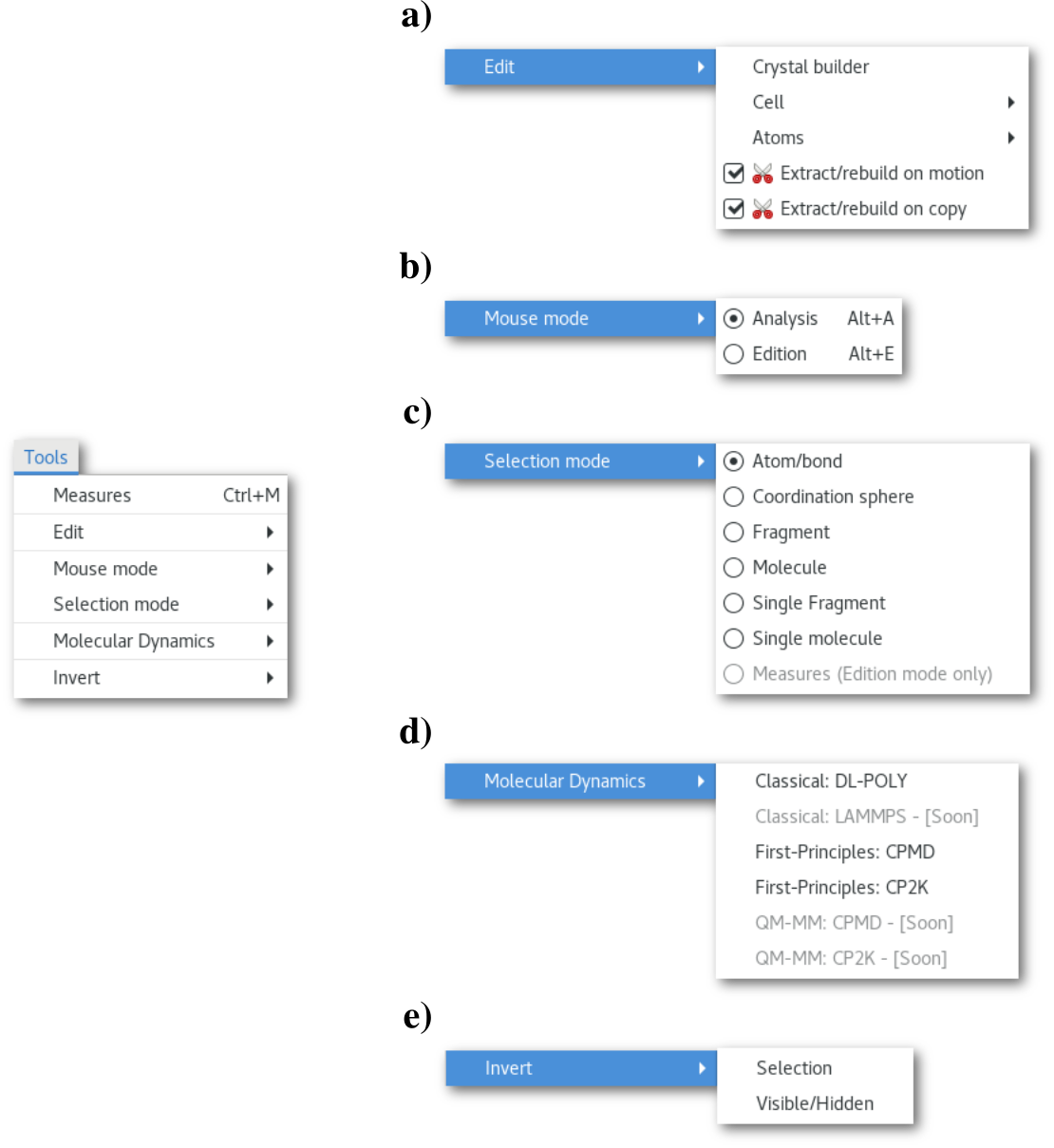
The "Edit" menu a), that regroups the edition tools will be covered in chapter 3.6, and the "Molecular Dynamics" menu d), regrouping the MD input assistants will be covered in chapter 7. Also the actions of the "Invert" menu e) being pretty intuitive, this section will only present the "Measures" button/dialog, the "Mouse mode" b), "Selection mode" c) menus.
The "Measures" dialog
Pressing the "Measures" button will open the "Measures" dialog [Fig. 5.10], that contains a notebook with 3 tabs, for inter-atomic distances, angles and dihedrals, and a button labelled "Font and style" to configure how the measure(s) will appear on the model.
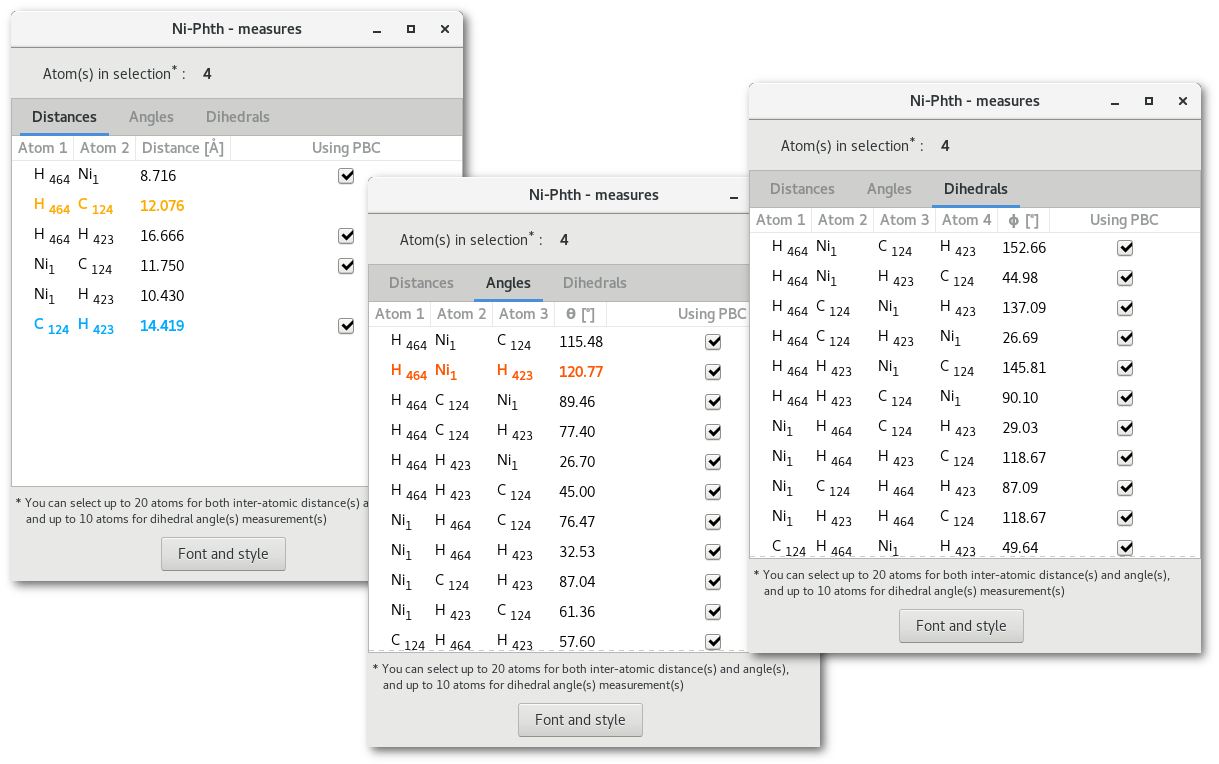
Each tab contains the respective measured properties for all the atoms in selection (see section 5.2 for more information on the selection process), this means the atoms that have been selected before opening the dialog, or, while it is opened and the tabs will refresh. The tabs read as follow:
First (2/3/4) columns the atoms id's
Column (3/4/5): the value measured in the model
Column (4/5/9): If PBC are used/required to measure the value (to avoid visual deception)
Behavior of the "Measures" dialog:
If too many atoms are in selection then the tabs will remain empty and the measures will not show up. To get the information about the measures back simply decrease the number of atoms selected under 20 atoms for the distances and the angles, and under 10 for the dihedrals.
If any line is selected by a mouse click in the tabs, then the font color of this line will change and the corresponding measure will appear in the model with the same color. In figure 5.10, 2 lines are selected in the "
Distances" tab, and 1 line is selected in the "Angles" tab, the result is presented in figure 5.11.
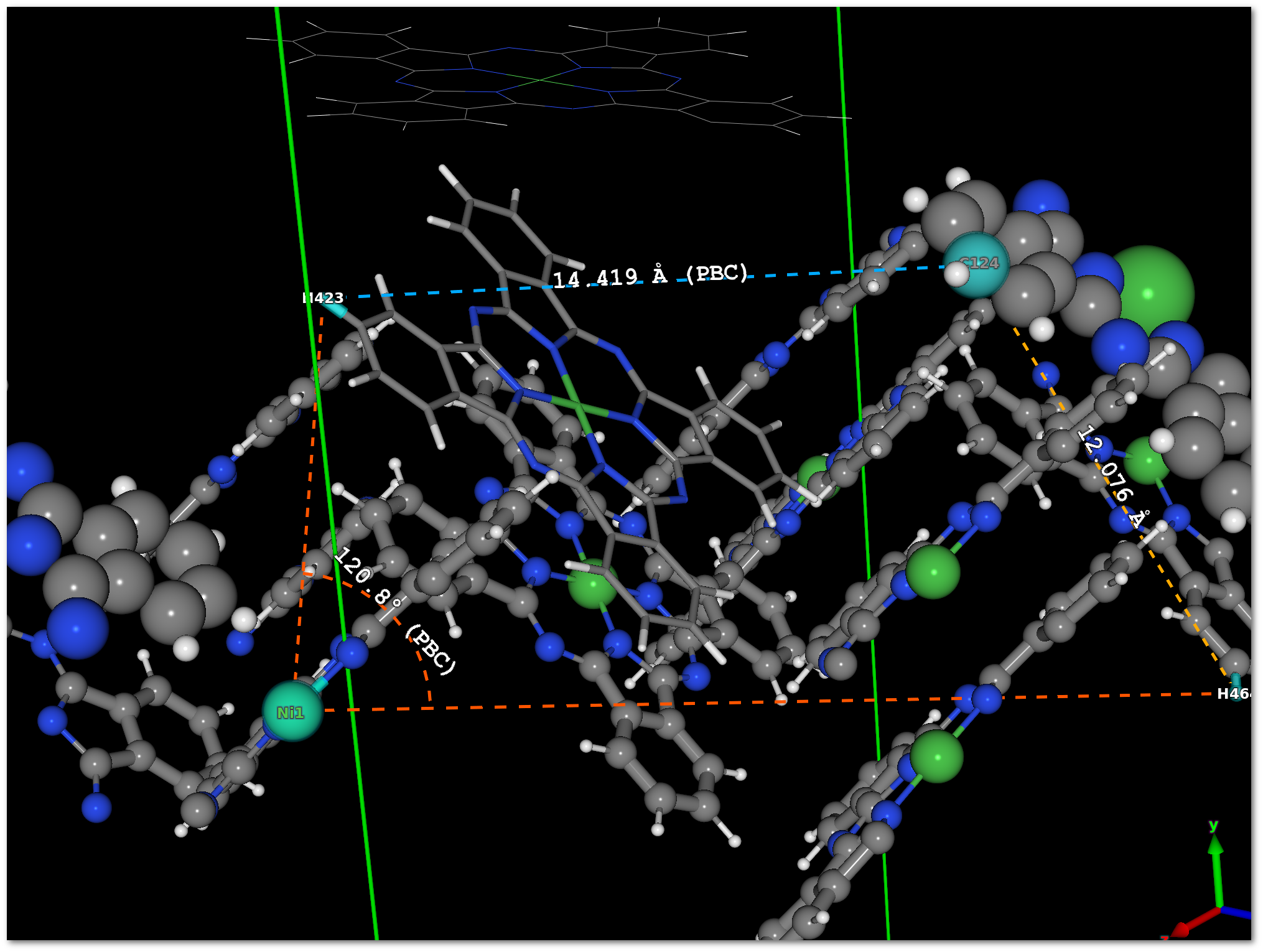
No visual information is available for the dihedral angles and only the numerical values are provided.
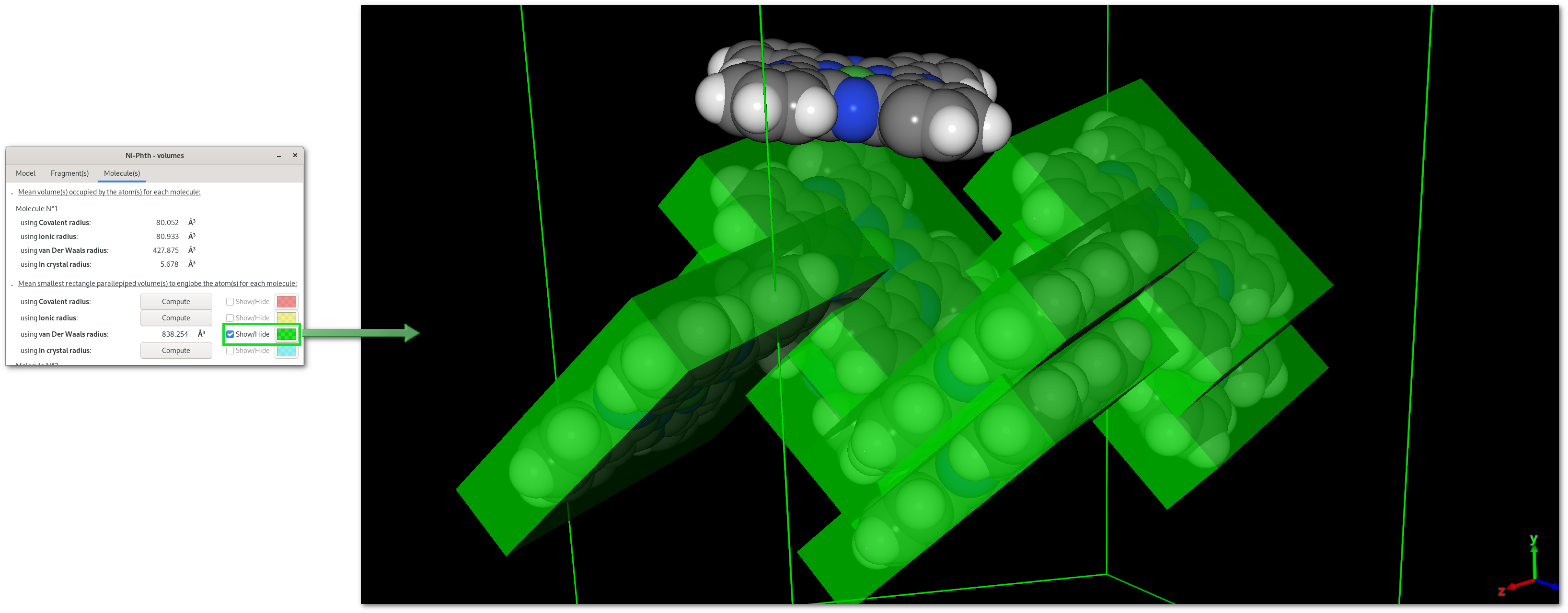
The "Volumes" dialog
Pressing the "Volumes" button will open the "Volumes" dialog [Fig. 5.12], that contains a notebook with 3 tabs, for overall atomic volumes ("Model"), isolated fragment volumes ("Fragment(s)"), molecular volume ("Molecule(s)").
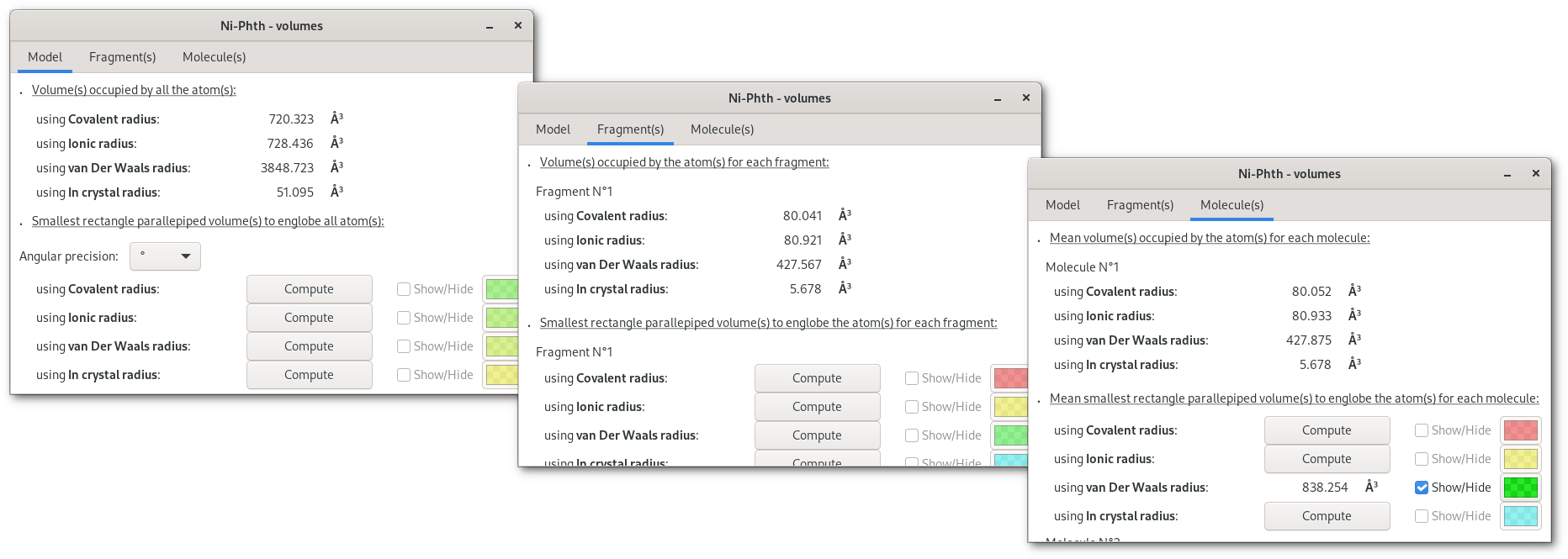
Tabs are divided in two parts: top for molecular volume(s) calculated for the respective target, bottom for calculations to dermine the smallest rectangle parallepiped volume for the respective target. Also volumes can be evalutated for different types of atomic radii (covalent, ionic, van Der Waals and in crystal), and on the first tab ("Volumes"), the angular precision (rotation of the volume) can be fine tuned for all calculations.
Once a "Compute" button is pressed the selected calculation will be perfomed, then numerical results become available and can be visualized pressing the corresponding "Show/Hide" button:
The "Mouse mode" menu
This menu allows to switch the mouse mode between "Analysis" (default) and "Edition". This menu is active, only and only if, there is a single configuration in the project (and not a molecular dynamics trajectory). Therefore when studying a MD trajectory only the "Analysis" mode is available.
The "Selection mode" menu
This menu allows to adjust the mouse selection capabilities, and switch between:
Atom/bond (default): selection atom by atom or bond by bond.
Coordination sphere: the atom and all its first neighbors.
Fragment: the atom and all other atom(s) that belong to the same fragment.
Molecule: the atom and all other atom(s) that belong to the same molecule.
Single fragment: same as [sfrag] and unselect all other atoms in the model.
Single molecule: same as [smol] and unselect all other atoms in the model.
Measures (Edition mode only): will be covered in chapter 6.