The Crystal builder window
The "Crystal builder" button [Fig. 6.2] allows to open the corresponding window.
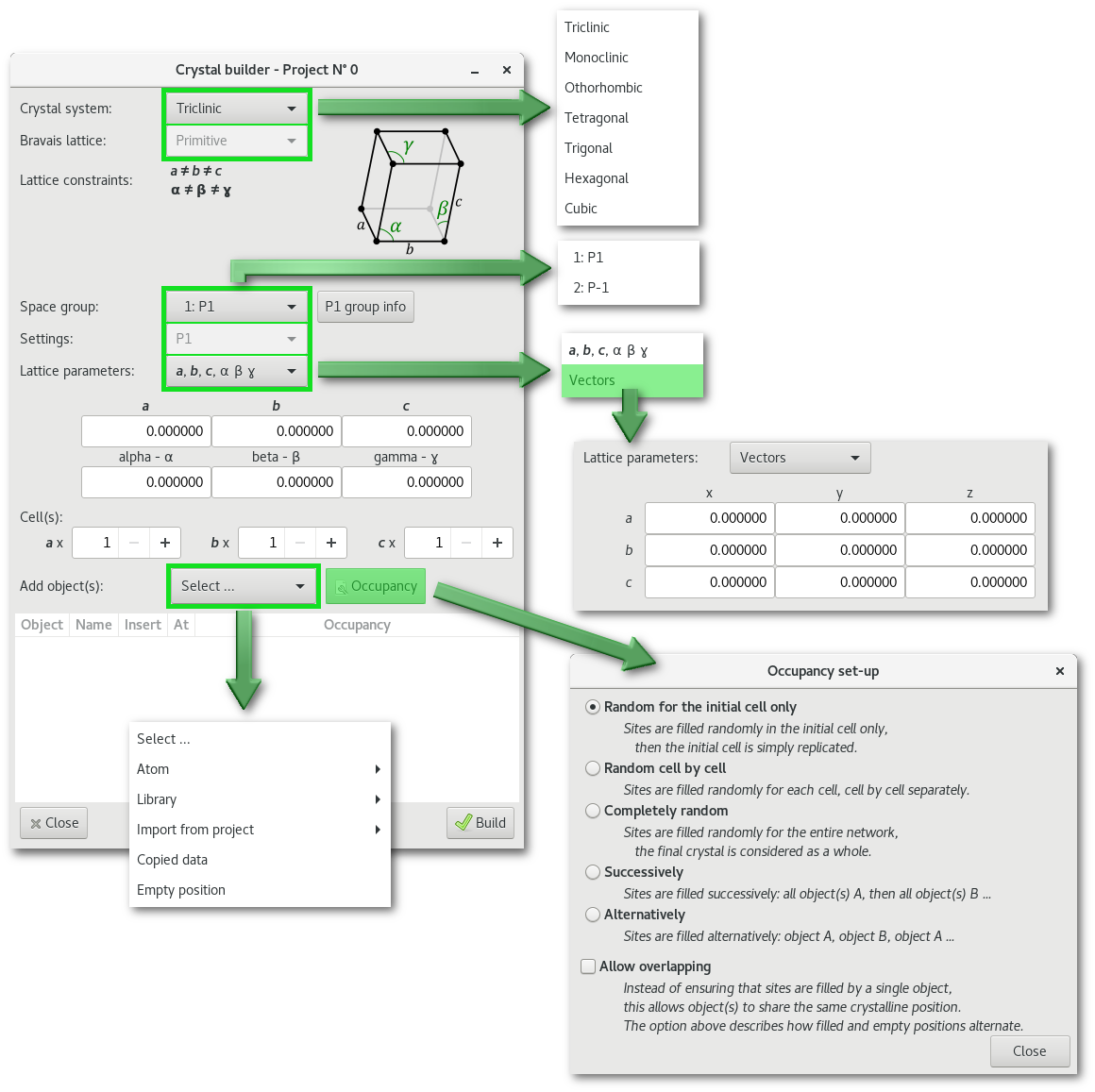
The "Crystal builder" window [Fig. 6.3] allows the creation of crystalline structure(s) and super-structure(s). Thus not only atom(s) but also molecules can be inserted to the crystalline positions defined by the space group.
The "Select ..." menu allows to insert objects: atoms, molecules, and atom selections from any other project opened in the Atomes workspace. It will be presented in details in section 6.1.2.10, at the exception of the "Empty site" button unique to the "Crystal builder" and that will be introduced in the next pages related to the occupancy.
Each object will be inserted at a position specified using the fractional coordinates, in the proportion of the occupancy, and following the rules of the selected space group. The following parameters can be adjusted:
The crystal system:
Triclinic
Monoclinic
Orthorhombic
Tetragonal
Trigonal
Hexagonal
Cubic
The type of Bravais lattice, depending on [cs], among:
Primitive
Base-centered
Body-centered
Face-centered
Hexagonal axes
Rhombohedral axes
The space group, from 230 groups of the International tables for Crystallography Vol. A, [1] and filtered using [cs] and [bv]
The space group setting, if more than one is available.
The format of the lattice parameters:
a, b, c and
Vector components: a(x,y,z), b(x,y,z), c(x,y,z)
The number of cell(s) to replicate on a, b and c to create the final supercell.
The object to insert to build the crystal using information specified at steps [cs], [bv], [sp], [sps] and [lp], including the following options:
The type of object (atom, molecule ...)
The fractional coordinates of the object
The occupancy of the crystallographic site (between 0.0 and 1.0)
How to handle occupancy (if < 1.0), object(s) and/or empty position(s):
"
Random for the initial cell only": sites are filled randomly in the initial cell only, then the initial cell is simply replicated."
Random cell by cell": sites are filled randomly for each cell, cell by cell separately."
Completely random": sites are filled randomly for the entire network, the final crystal is considered as whole."
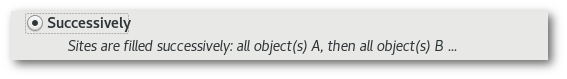
Successively": sites are filled successively, all object(s) A are inserted (for the first"
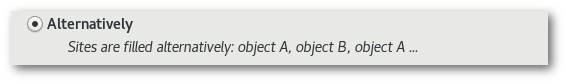
Alternatively": sites are filled alternatively: object A is inserted on the first position, object B is inserted on the second position, object A on the third position, object B on the fourth position ... and so on.
In any case the number
Overlapping, object(s) on the same position, can also be allowed, the concept will be illustrated in the following examples.
Using the crystal builder: space group information
Once a space group has been selected it is possible to access the "Space group info" dialog using the "group info" button:
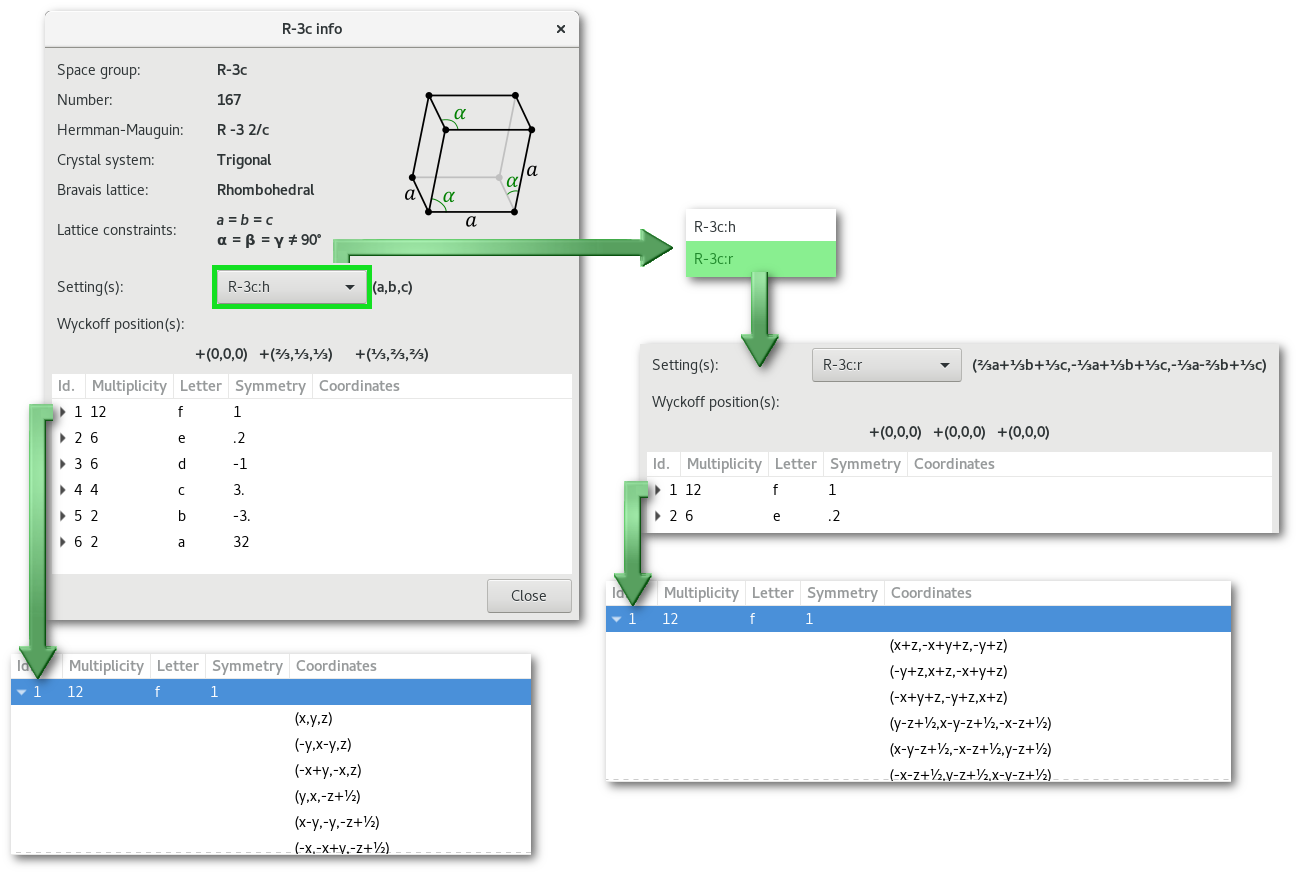
The main information available is presented in the "space group info" dialog [Fig. 6.4], including the different setting(s), and the corresponding initial coordinates and Wyckoff positions.
Using the crystal builder: building crystal(s)
The following pages will present crystal building examples using the Fd
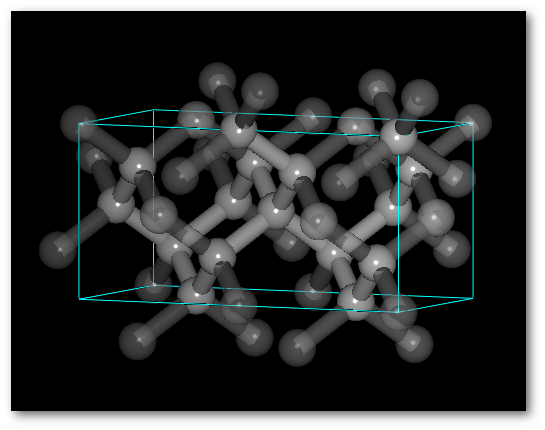
The C-diamond structure:
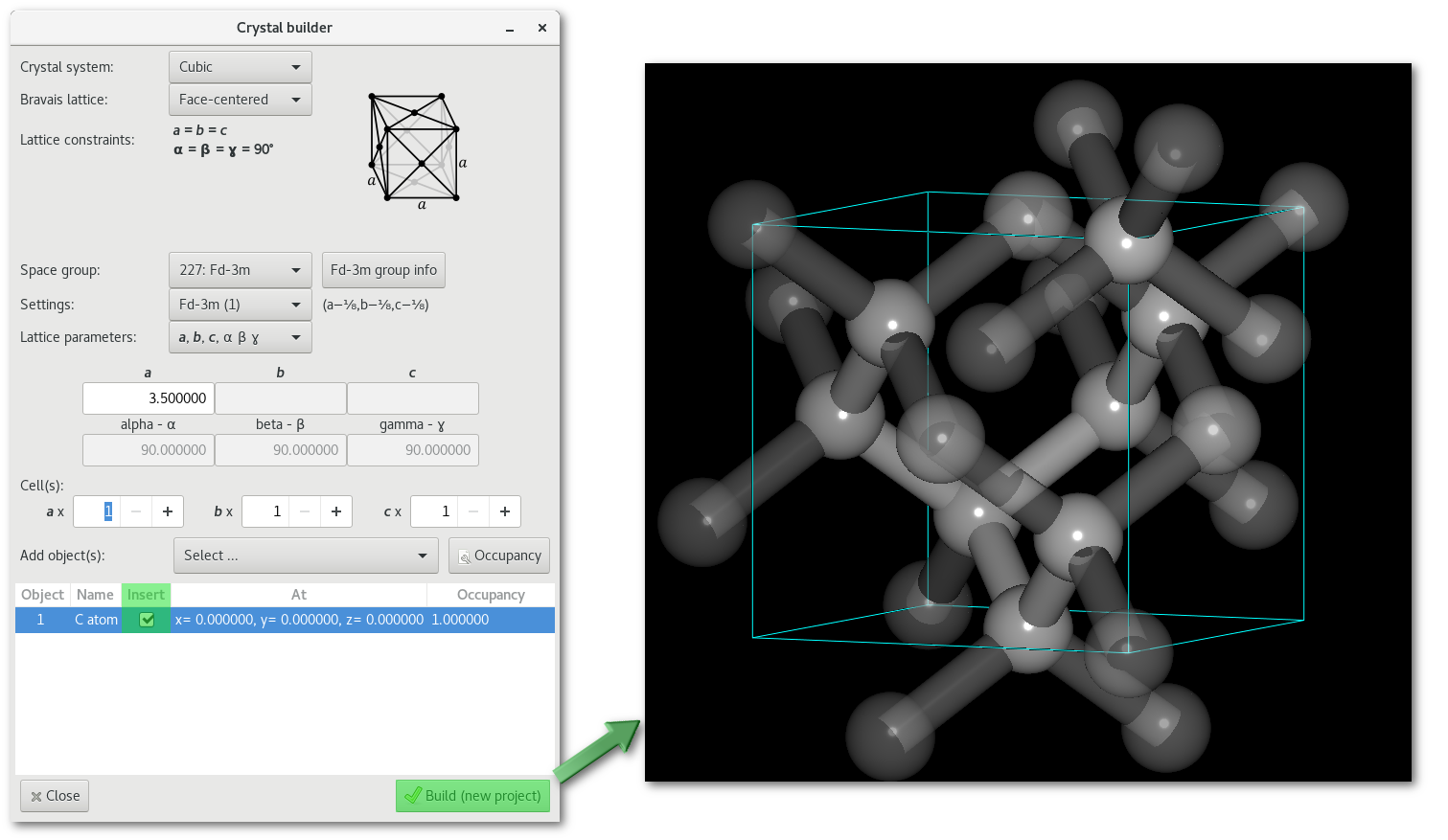
The following steps are required to build the C-diamond crystalline structure (see figure 6.5-left):
Select the Fd
Cubic -> Face-centered -> 227: Fd-3m")Set the value for the lattice parameter:
Insert a C atom at (0.0, 0.0, 0.0) with occupancy to 1.0, you can edit both coordinates and occupancy by double-clicking on the corresponding line.
Select the "
Insert check button" to use the atom to build the crystal, or alternatively click to top of the "Insert" column to insert all elements.Finally simply click on the "
Build / Build (new project)" button.
The crystal is then displayed (see figure 6.5-right), also clones (atoms linked using the PBC see section 5.1.2.3) are immediately shown. The existence of a chemical bond depending on the cutoff(s), determined automatically when creating the crystal, you might need to adjust the values to define properly the bonding of the system.
The C-diamond structure, playing with occupancy:
In these examples the parameters to build the crystal remain exactly similar to [cd1], only the total number of cells will be increased to 2 on a: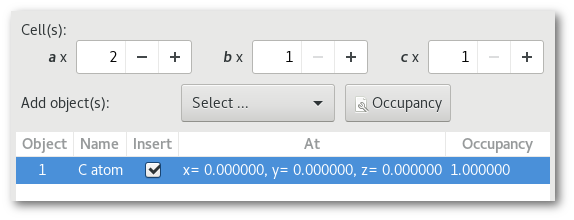
Occupancy = 1.0:
With 8 atoms per cell, the total number of atoms in the system is equal to 16.
Occupancy = 0.5:
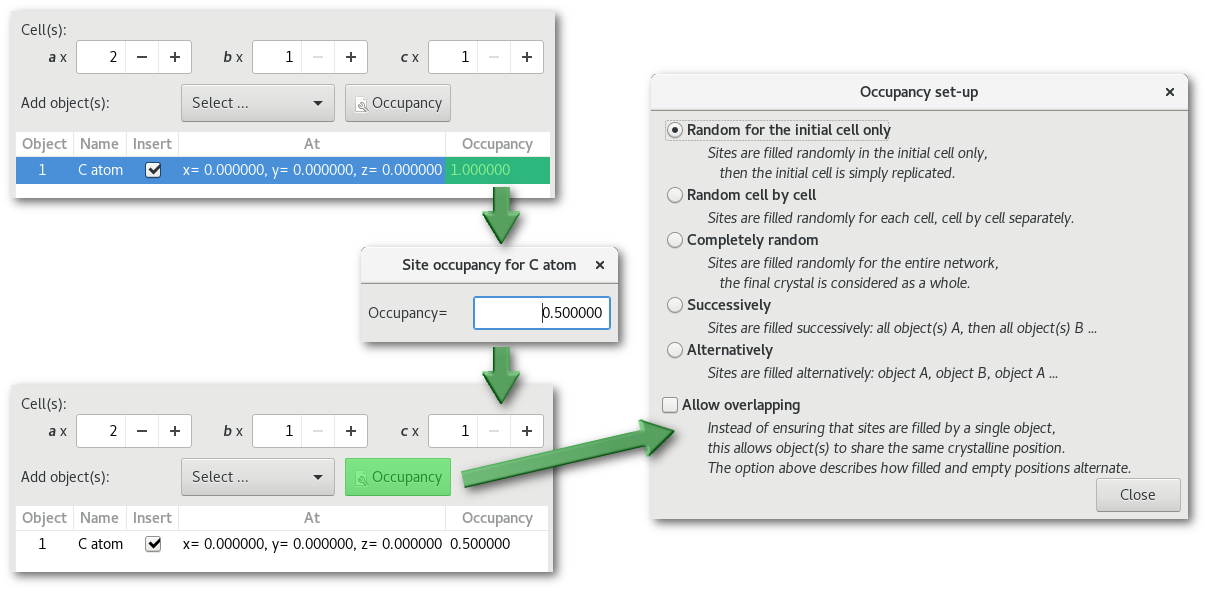
With an occupancy of 0.5 the number of atoms per cell will be reduced to 4,
and the total number of atoms in the system to 8.
(1) 
"
Random for the initial cell only":
The same treatment is applied to each cell, inducing an overall symmetry.
The system is not disordered with 4 atoms per cell at the exact same positions.
(2) 
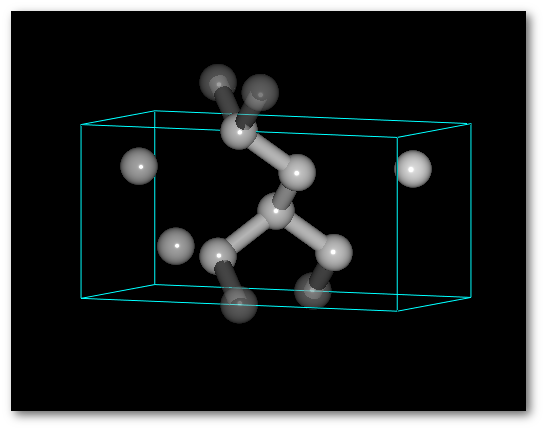
"
Random cell by cell":
A random treatment is applied to each cell independently.
The system is disordered, but the number of atoms per cell remains equal to 4.
(3) 
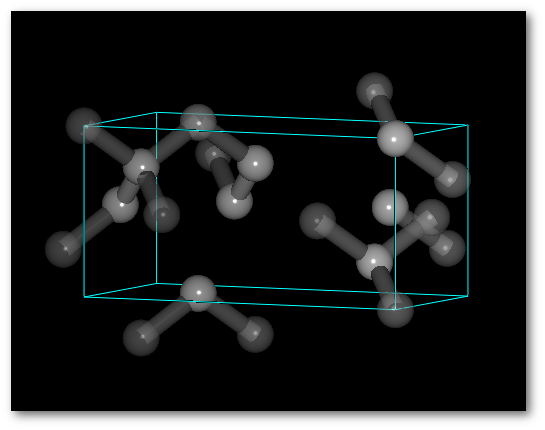
"
Completely random":
The system is entirely disordered.
The average value 0.5 C atoms per site is respected for the entire structure,
but the number of atom(s) per cell can change.
This option if particularly useful when the occupancy for a particular site is very low, see the section remarks afterwards for more information.
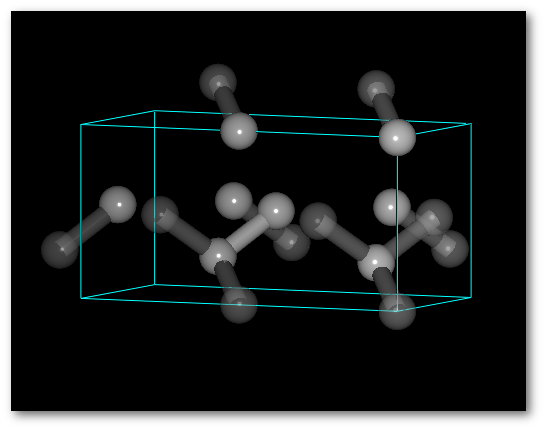
To illustrate (4) "Successively" and (5) "Alternatively" it is interesting to use more than one chemical species:
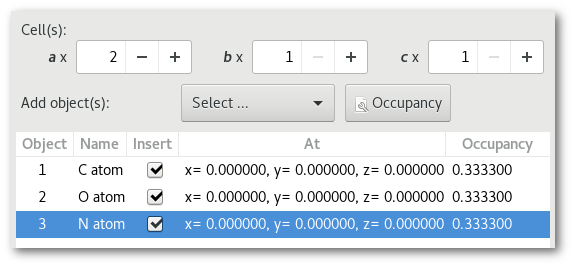
Adding O atoms: Adding O and N atoms: For all chemical species: For all chemical species: - The occupancy is equal to 0.5 - The occupancy is equal 0.333 - The site is the same (0.0, 0.0, 0.0) - The site is the same (0.0, 0.0, 0.0) 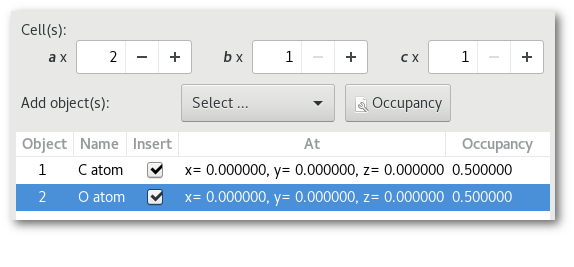
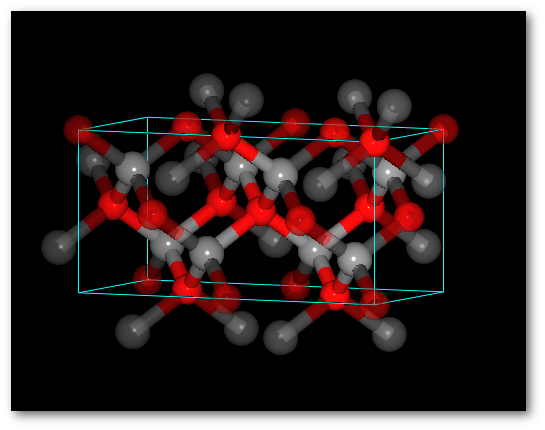
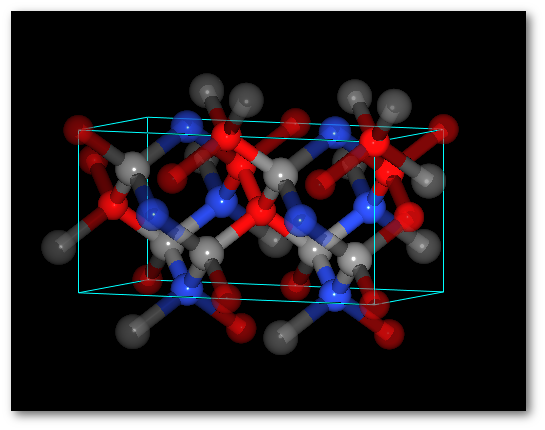
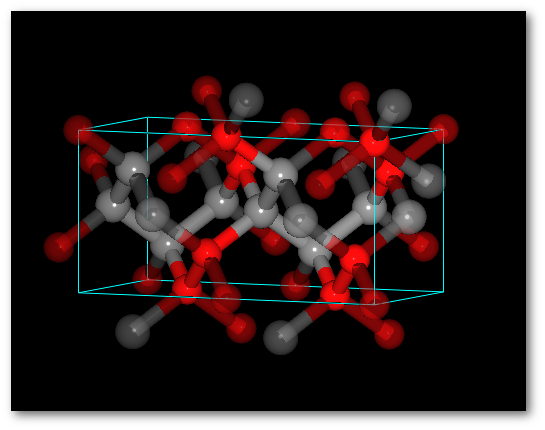
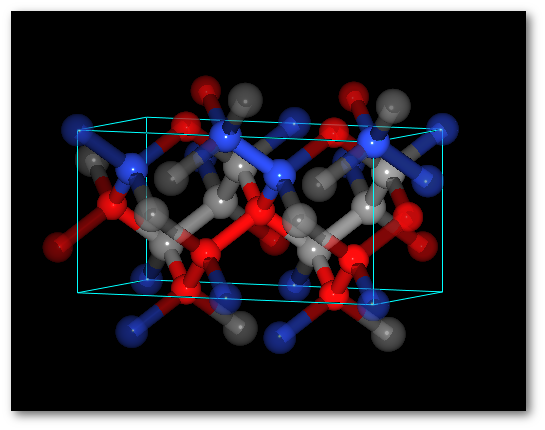
In both cases (4) "Successively" and (5) "Alternatively":
The system is ordered.
The average values of the occupancy per site are respected for the entire structure.The C-diamond structure, playing with molecules:
As already mentioned Atomes does not only allow to build atomic crystalline structures but also molecular crystalline super-structures.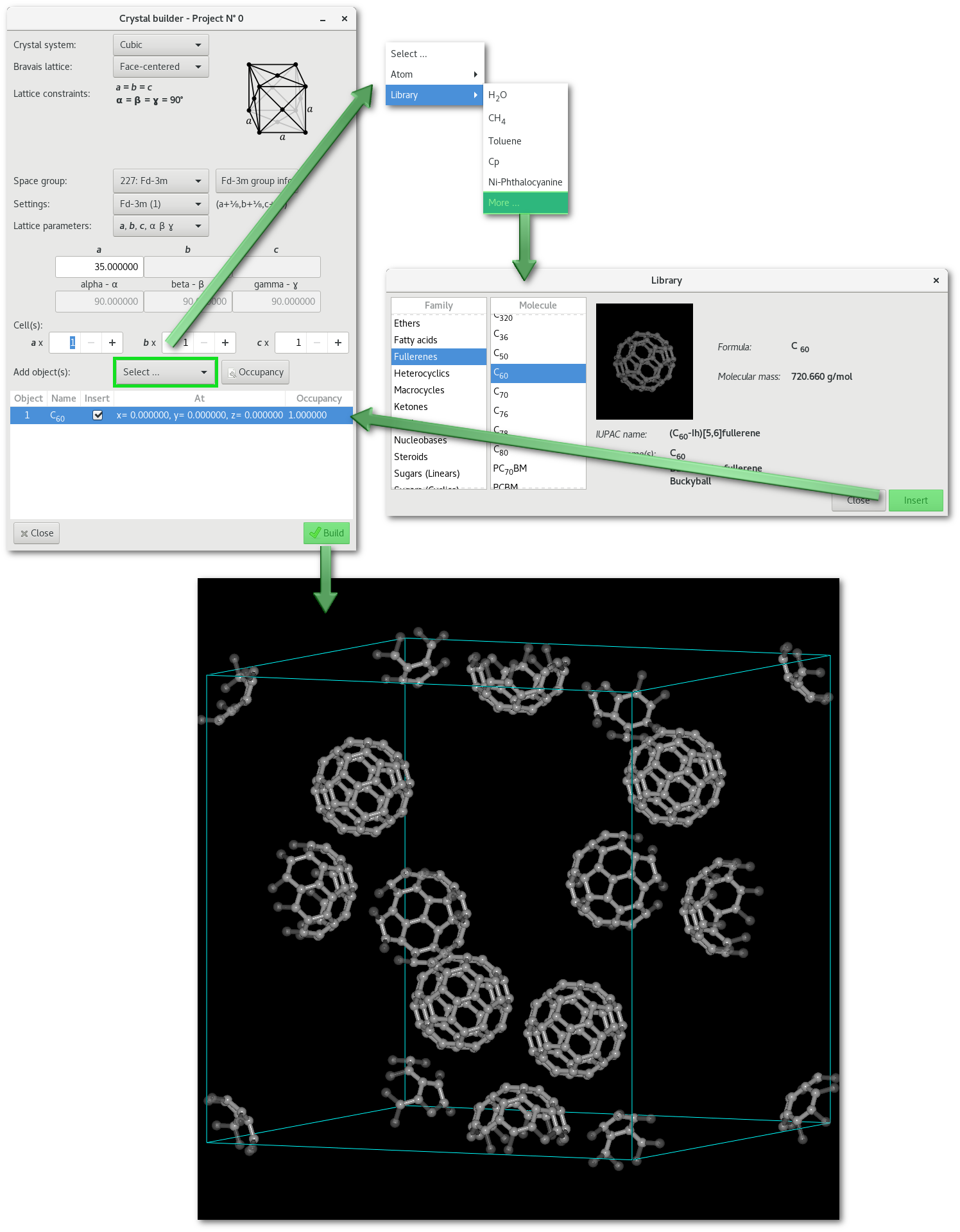
The "
Insert..." menu allows indeed to insert fragments from the library or from any project opened in the workspace.
Using C
As for the atom(s) tweaking the occupancy can be useful with molecules: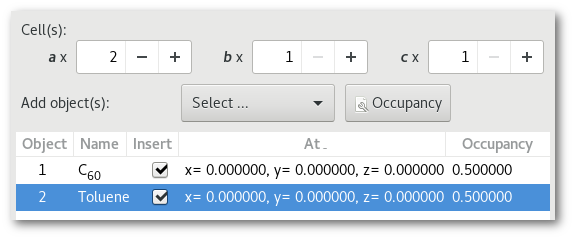
Using the (4) "
Successively" occupancy set-up, you build: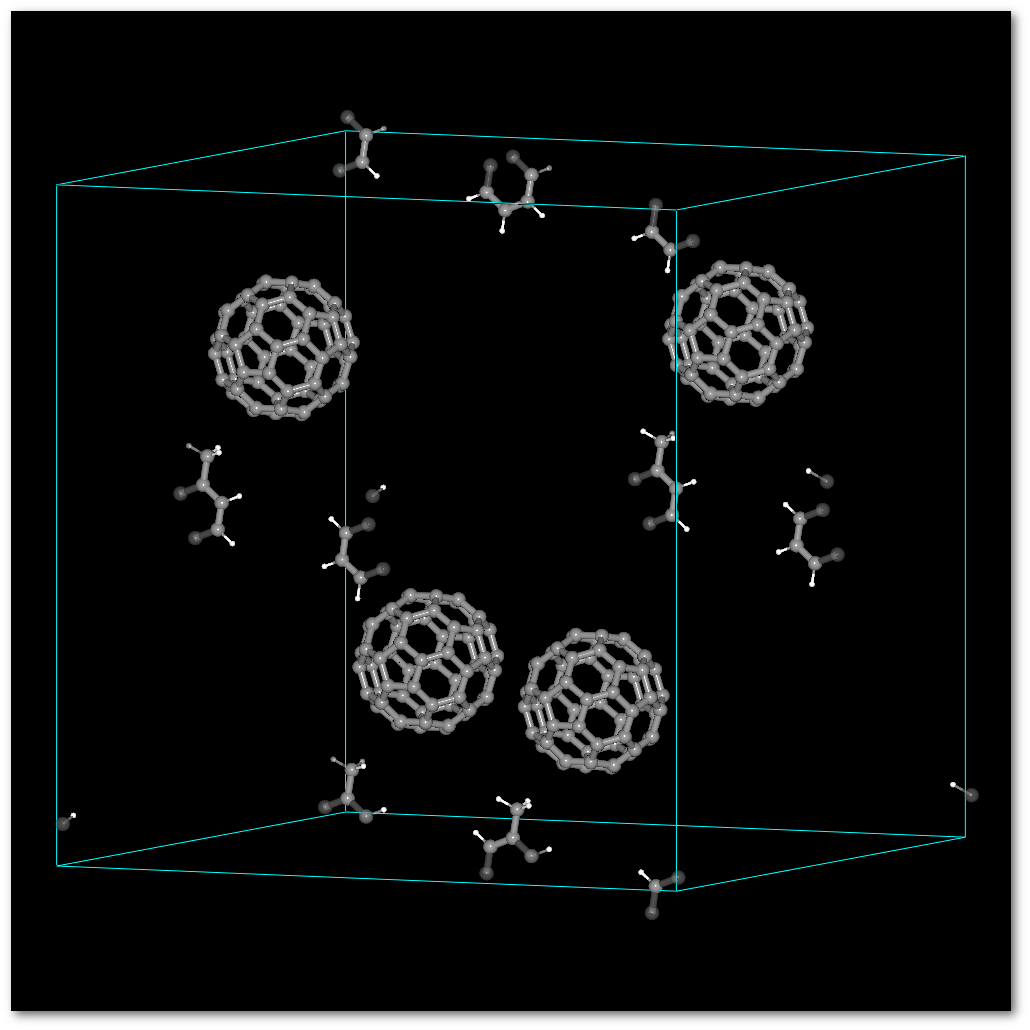
The C-diamond structure, molecules and overlapping:
As mentioned using the "Allow overlapping" option allows to insert at the same crystalline positions. The interest is limited to molecules-molecules overlapping or molecules-atoms overlapping, there is no point in getting two atoms at the same position. The idea behind this concept is to allows the encapsulation of an object by another one: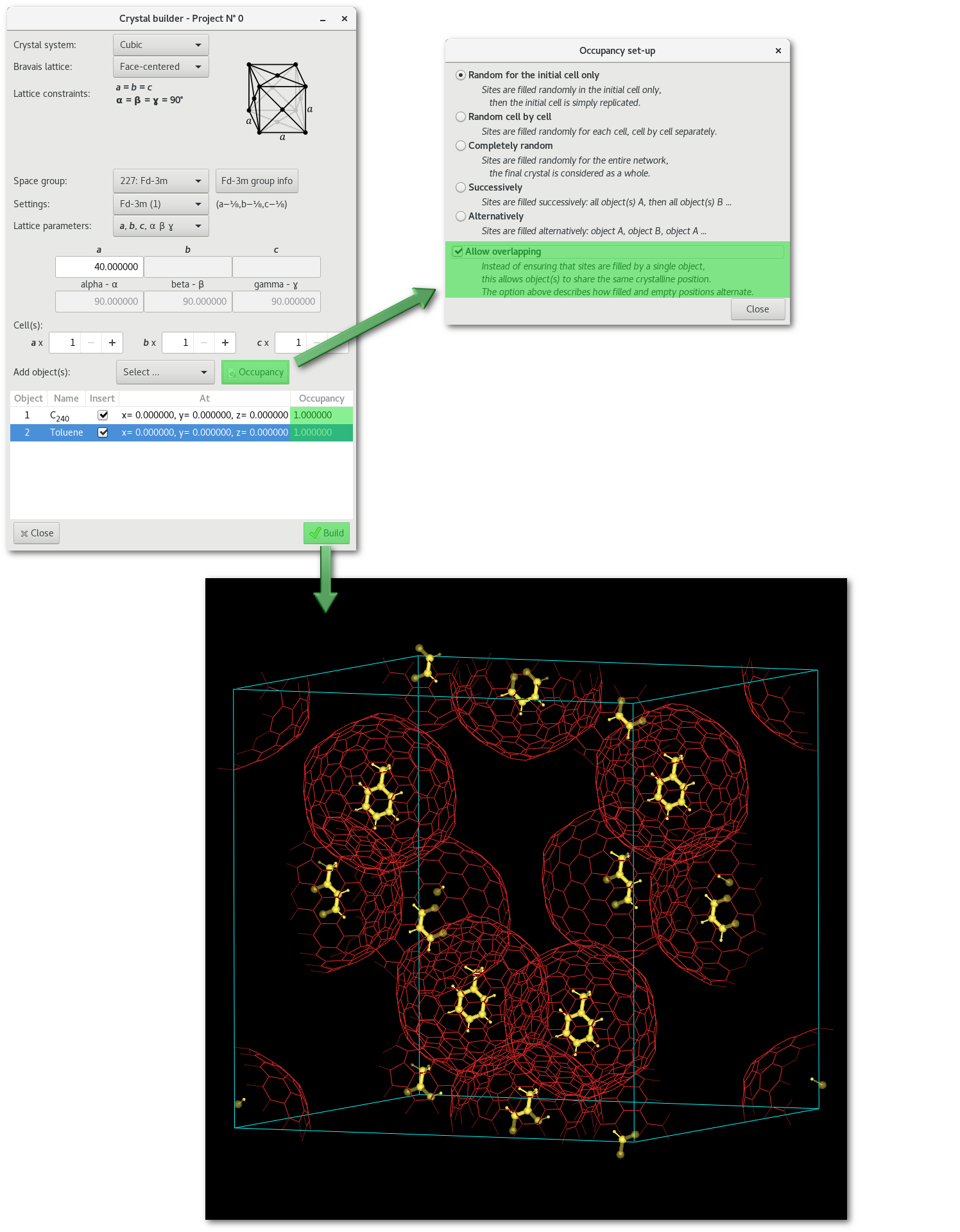
Crystal building: for more on the crystal building process see appendix 4.
Here are some important things happening when building a crystal in Atomes:At the beginning of the process the size of the object(s) to insert is compared to the lattice parameters, if the lattice parameters are considered too small a warning message will pop-up:
For an atom, the size is set to 1.0 Å.
For a molecule, the size is the maximum interatomic distance within the molecule.
The process is not fail-safe, and without being careful it is possible to build a crystal with an improper bonding.
The distance matrix calculation to determine bonding information is performed on the fly at the end of the building process, if the crystal bonding is too bad, this calculation could fail and no bond will be display. If the crystalline structure is good, then simply correct the bond cutoff to perform the calculation again, and get the correct bonding information.At the end of the process all the atoms are wrapped back in the unit cell.
The bond cutoff are determined automatically, therefore the visual aspect of the final structure might be misleading, with too much of too few bonds compared to what was expected.
When inserting a molecule remember that there is not way to determine its orientation, with water molecules for instance it might be required to rotate the molecule(s) afterwards, see section 6.1.3 for more information.
Occupancy: as illustrated previously the order the object(s) is(are) inserted on the crystalline positions might have an importance on the final structure. This is true when the occupancy is < 1.0 and / or when object can share the same site when using the "
Allow overlapping" option.
To ensure that the desired crystal will be built, either insert the object in the proper order or re-order them using the mouse drag an drop: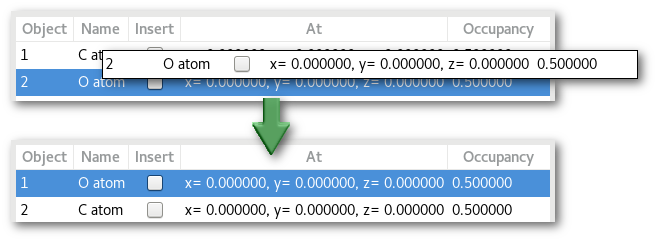
The occupancy
Empty position: the "
Empty position" button of the "Select ..." menu is used to force the insertion of empty position(s).
The empty site(s) are always treated before any object insertion(s), even if the position in the insertion tree is not on top.
In the case of "Overlapping"+"Empty position" the rules for occupancy are modified as follow:
- "Space-group symmetry," International Tables for Crystallography, vol. A, 2016.