The Model edition window
The "Model edition" window offers several tools dedicated to the edition of the atomic coordinates and is accessible using any of the buttons of the "Atom" submenu [Fig. 6.11].
It is not possible to access the "Model edition", and therefore to move / replace / remove and insert atom(s) in the case of MD trajectory.
Selection process
When the "Atom edition" window is opened the main OpenGL window remains active, and it is still possible to work in it. However having the "Model edition" window opened will modify the results of the atom selection process, whether is it performed using any dialog window or using with the mouse describe in section 5.2.1 and 5.2.2. The newly extra-selected atom(s) will be covered in pink (instead of light blue), see [Fig. 6.12], and even already selected atoms (in light blue) can be re-selected.
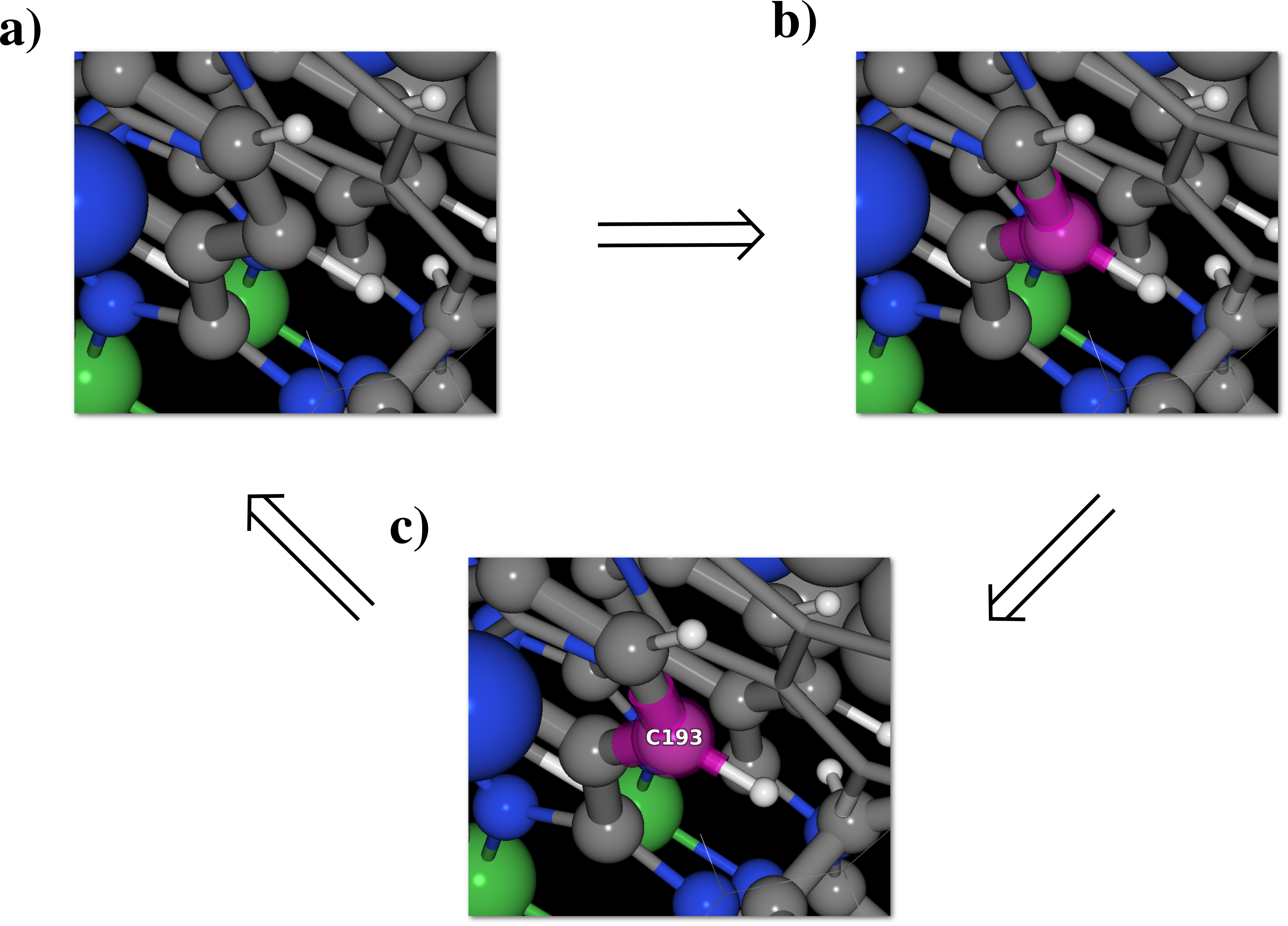
As long as the "Model edition" window remains open, the "Measures" dialog [Fig. 5.10] will only presented measurement(s) related to the new pink atoms.
The utilization and purpose of this extra selection feature will be illustrated when presenting the action tabs of the "Model edition" window.
General behavior
Before browsing each and every tab of the "Model edition" window it is required to introduce the "atom search" tool [Fig. 6.13] common to almost every tab (in the top part) in the window. Each tab being dedicated to a particular action the search tool will help to find and selected the atom(s) you want the action to be performed upon:
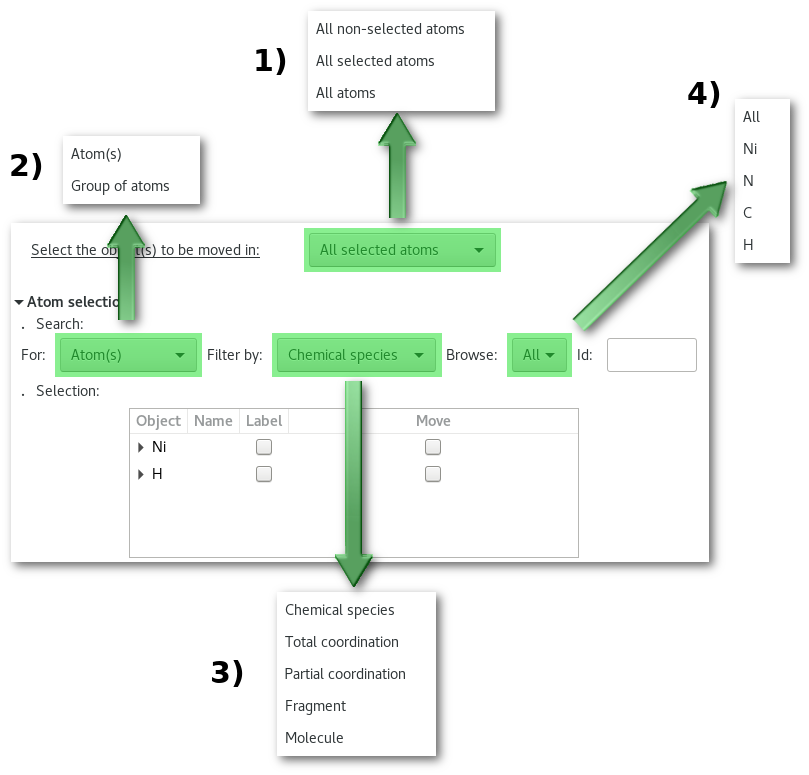
Search for atom(s) among [Fig. 6.13-1]:
non-selected atom(s) only: search only in "
All non-selected atoms" [Fig. 6.13-1]selected atom(s) only: search only in "
All selected atoms" [Fig. 6.13-1]or all atoms: search in all "
All atom(s)" [Fig. 6.13-1]
[selao] is the default value for menu [Fig. 6.13-1] if some atoms are selected, and [allao] is the default value otherwise.
Apply the action to [Fig. 6.13-2]:
The atoms as single object(s): pick "
Atom(s)" [Fig. 6.13-2]Group of atom(s): pick "
Group of atoms" [Fig. 6.13-2]
In case [isoat] the action will be performed on the atom(s) that will be selected and each of these atoms will be treated as an isolated object. In case [groat] the action will be performed on the group(s) of atoms, the atom(s) that will be selected afterwards belong to: entire coordination sphere(s), fragment(s) or molecule(s).
Filter the result of the search depending on the type of object you are interested in figure 6.13-3:
Chemical species (Available only if [isoat] is selected for [Fig. 6.13-2])
Total coordination
Partial coordination
Fragment
Molecule
Pick the appropriate value in figure 6.13-3 and the selection tree bellow will be updated accordingly.
Refine the search to present only the atom(s) of that particular chemical species (if [isoat] was selected in figure 6.13-2) or the group of atoms that contains such chemical species (if [groat] was selected in figure 6.13-2)
Notes:
Clickable : some of the column headers in the search tree are clickable, in the top part if you click on the "
Label" or "Move" the corresponding action will be applied / unapplied to the entire data set available in the search tree.Update : whenever the values for the menus [for] [Fig. 6.13-2] and [fil] [Fig. 6.13-3] are modified the tree is not only refreshed but also cleaned, and so is the corresponding data thus previous selection sets would be lost in the process.
Examples of the utilization of the "atom search" tool [Fig. 6.13] will be presented along with each actions available in the "Model edition" window.
The "Move" tab
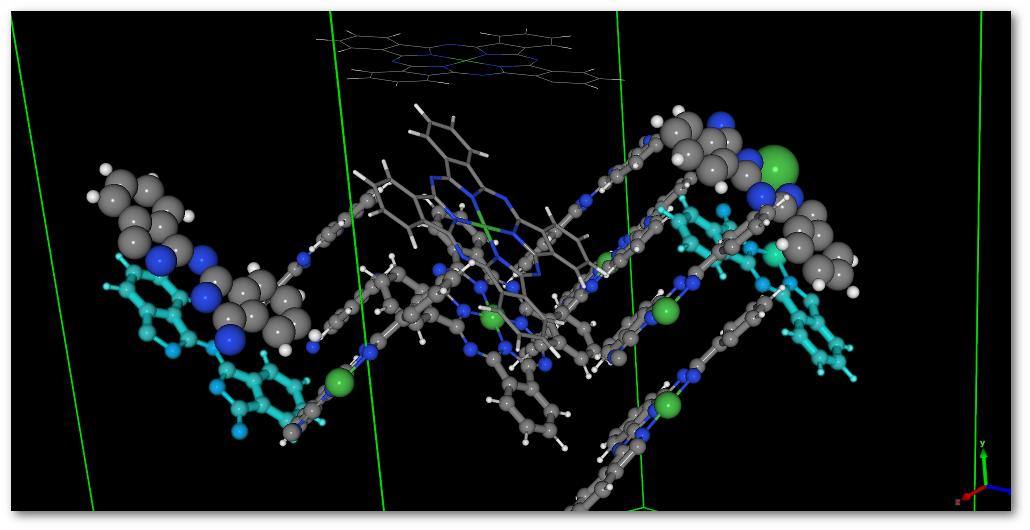
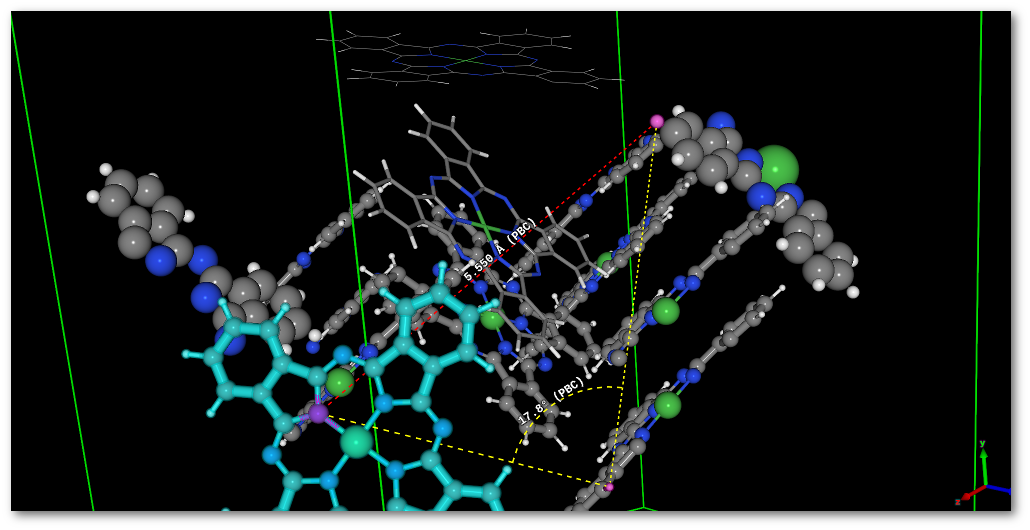
The "Move" tab allows to move atom(s) (translation only) or group of atoms (translation and rotation around the groups barycenter). In the following example one of the fragment in the model is selected, the fragment appears to be split because of the periodic boundary conditions:
To move the selected fragment:
Open the "
Move" atom(s) tab: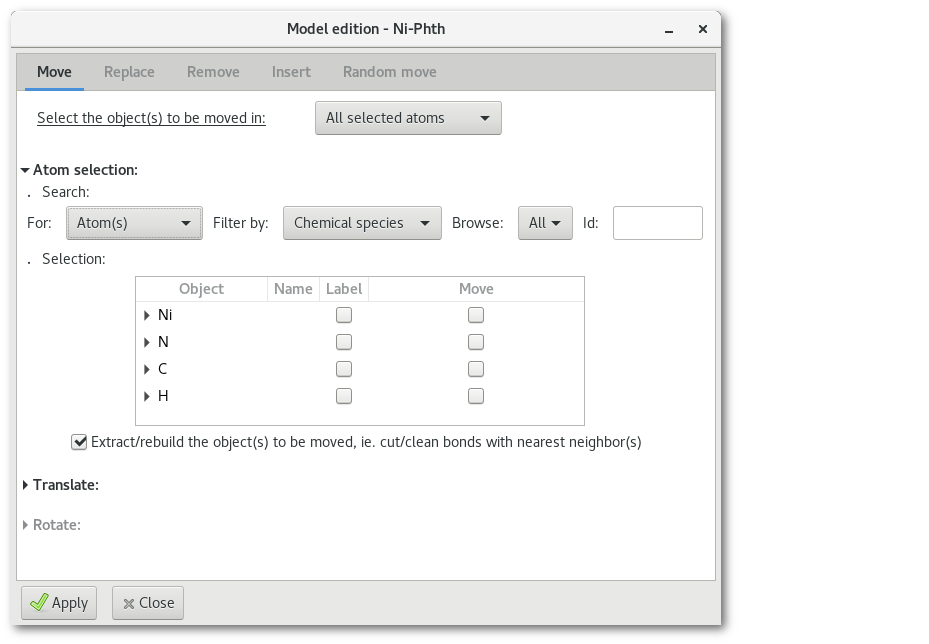
Go back to the main OpenGL window, keeping the "
Model edition" window opened, and select few atoms, that will appear in pink color. Then open the "Measures" dialog [Fig. 5.10] to select a bunch of measurements to be displayed: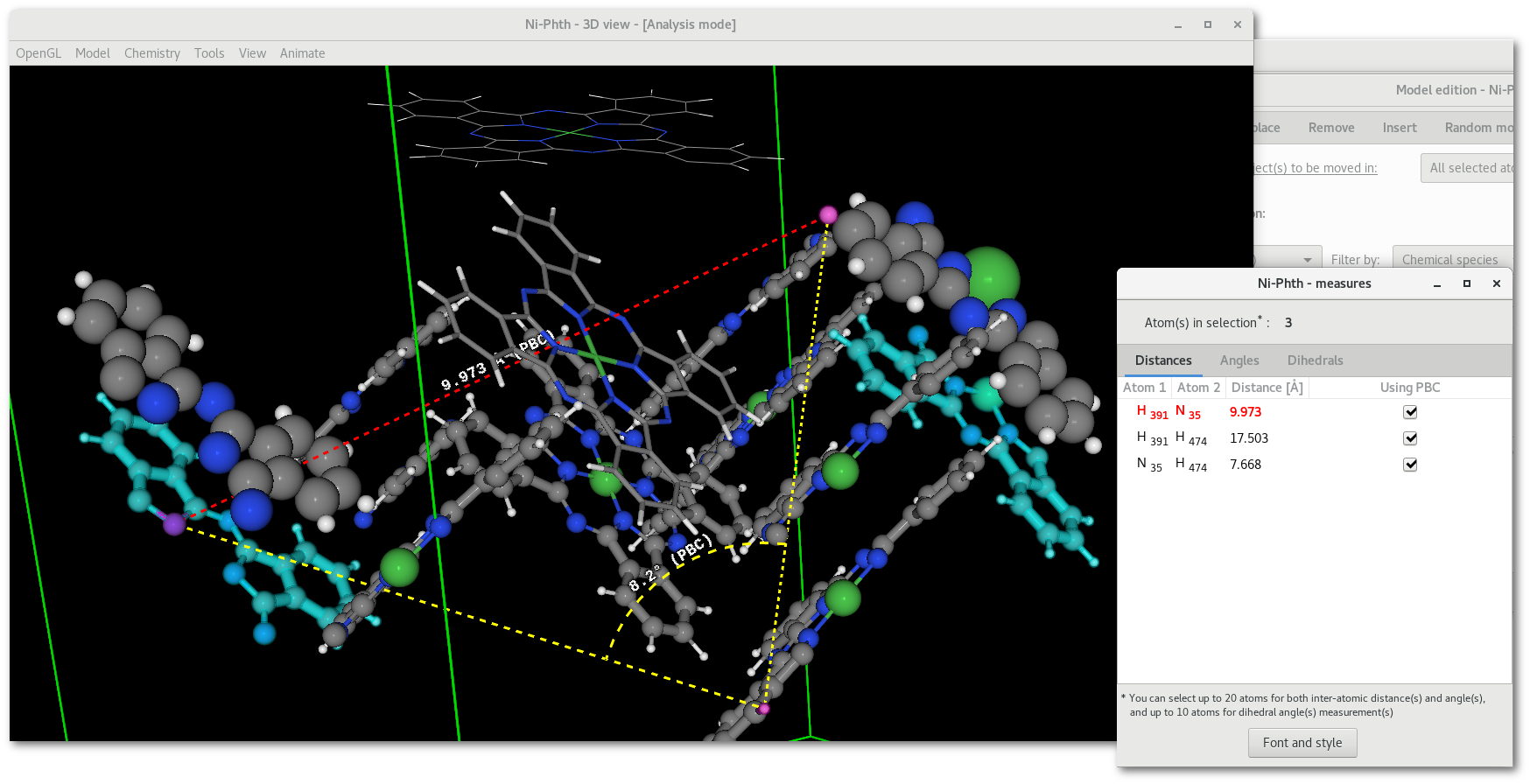
Go back to the "
Model edition" window and adjust the search "For" option and select "Group of atom(s)":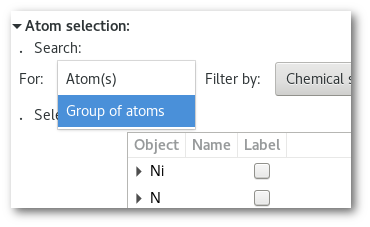
Adjust the search "
Filter by" option and select "Fragment":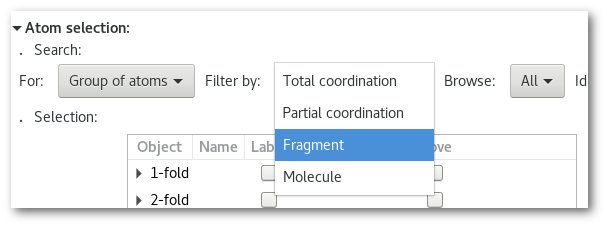
Then select the fragment in the selection tree:
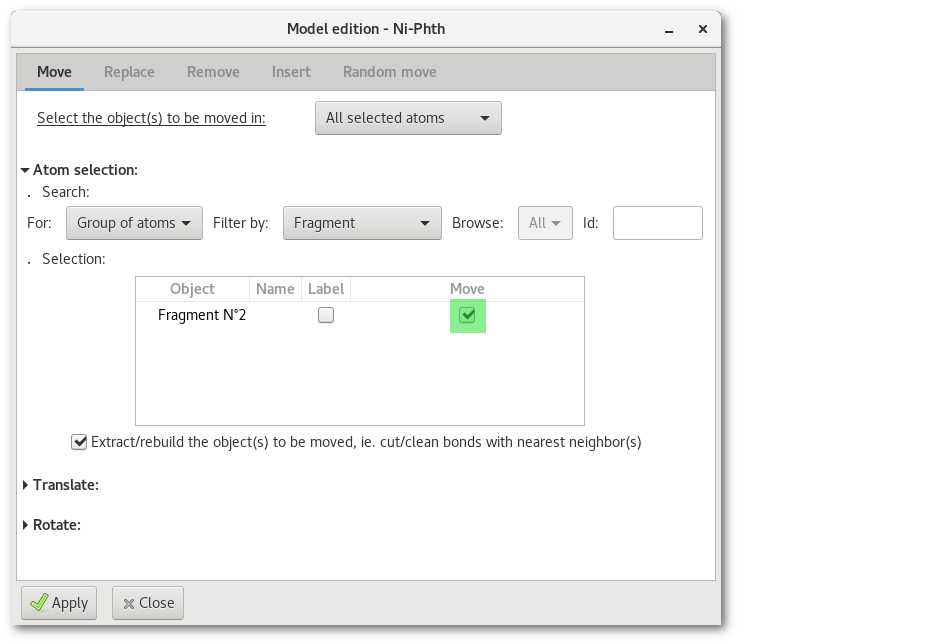
Open the lower part(s) to move the fragment:
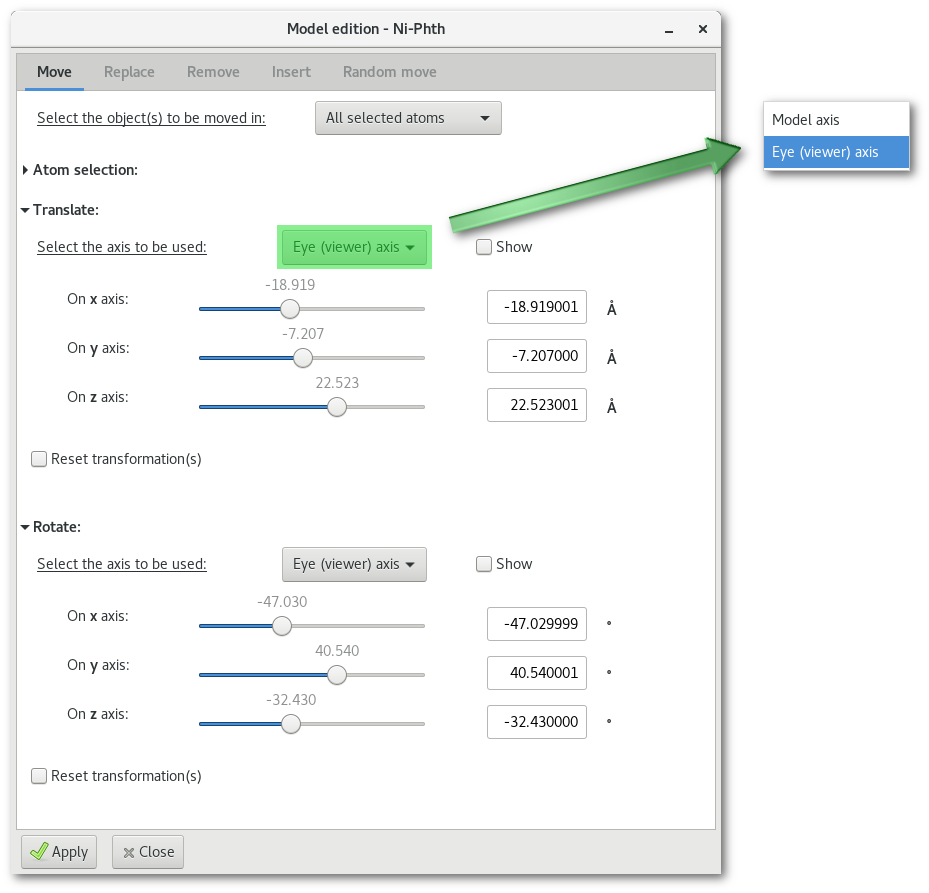
Motion axis: it is possible to select (and visualize) the motion axis and choose between the "
Eye (viewer) axis" and the "Model axis" (real x, y and z of the atoms).Motion actions: the motion interactors (ranges and input entries) allow to adjust motion very precisely, slightly more approximate motion is also available using the mouse (see Sec. 6.2)
Measurements: if already present in the OpenGL window, measurement(s) are updated on the fly, allowing to adjust perfectly position and orientation of the object in the model, see [Fig. 6.15].
Motions: translation is always available, however rotation is only available for "
Group of atom(s)" and is performed around the barycenter(s) of the object(s) to be rotated.Reconstruction: the check button labelled "
Extract/rebuild the object(s) to be moved, ie. cut/clean bonds with nearest neighbor(s)" offers the following option:For "
Atom(s)": if activated the object(s) to be moved will be extracted from the model and translated independently, otherwise chemical bond(s) will simply be stretched.For "
Group of atom(s)": if activated the atomic positions of the object(s) will be corrected to get only single piece object(s).
As illustrated in figure 6.14 the fragment visually appears in 2 pieces, and as illustrated by the example as a single piece object after motion [Fig. 6.15].
If the object(s) to be moved are coordination spheres and if the spheres that are being moved share atom(s), then the positions of these atoms might be affected twice by this procedure, leading to an awkward, yet correct, 3D representation.Bonding: During motion, the coordination information is modified, all related menus and corresponding dialogs are updated accordingly, however detailed bonding information is lost. It is therefore strongly recommended to recompute bonding properties afterwards.
The "Replace" tab
The "Replace" tab allows to replace object(s), atom(s) or group of atoms, by either: atom(s), molecules imported from the internal library or atom(s) imported from any project opened within the same instance of the Atomes program:
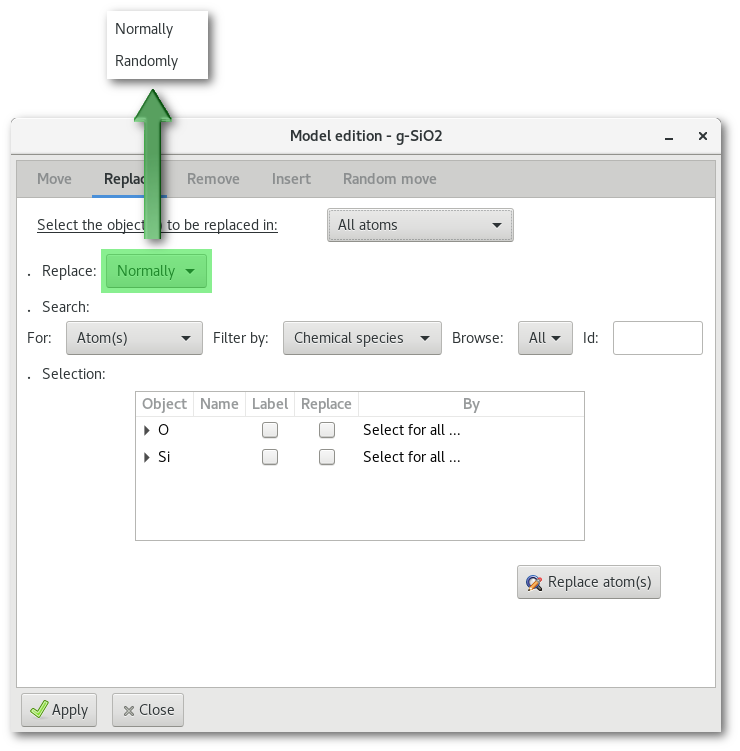
As illustrated in figure 6.16 it is possible to replace object(s) one by one ("Normally") or by random pick ("Randomly"). The difference between the normal and the random search trees are illustrated in top part of figure 6.17.
For normal substitution(s) [Fig. 6.17-a] it is possible to select the replacement object(s) either at once for all objects, using the "Select for all ..." menu, or alternatively one by one when browsing the tree and using the "Select ..." menu(s).
For random substitution(s) [Fig. 6.17-b] only a single replacement is selected for each type of object, and it is require to enter the number of substitution(s) to be performed.
The substitution options accessible using the "Select ..." menu are illustrated in the bottom part in figure 6.17 with the different "atoms" c), "Library" d) and "Import from project" e) submenus.
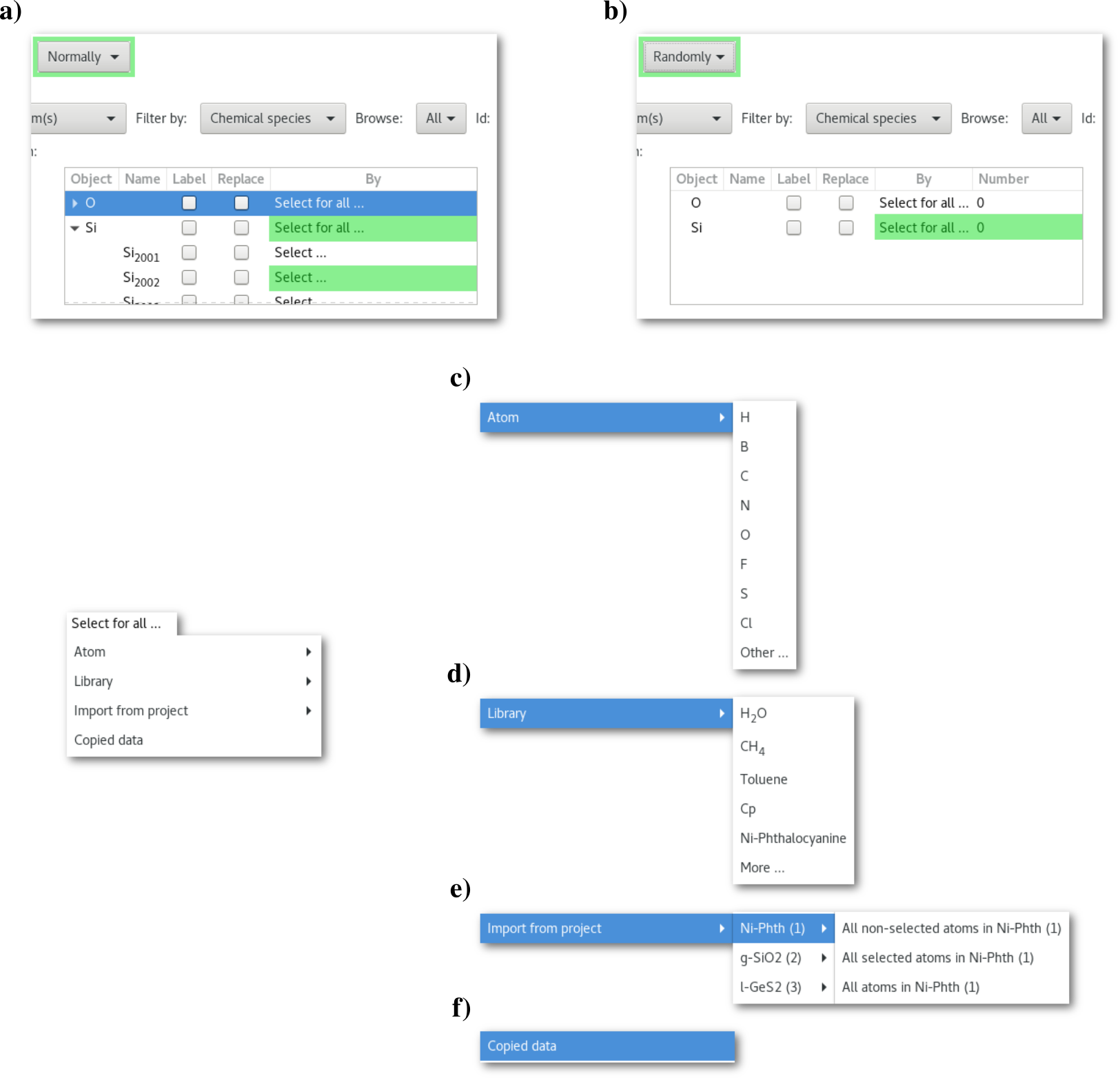
:
The "
Atom" submenu [Fig. 6.17-c], to insert a new atom in place of the object(s) to be removed. Some shortcuts are proposed, if required the "Other ..." button allows to open a periodic table to pick any appropriate atom: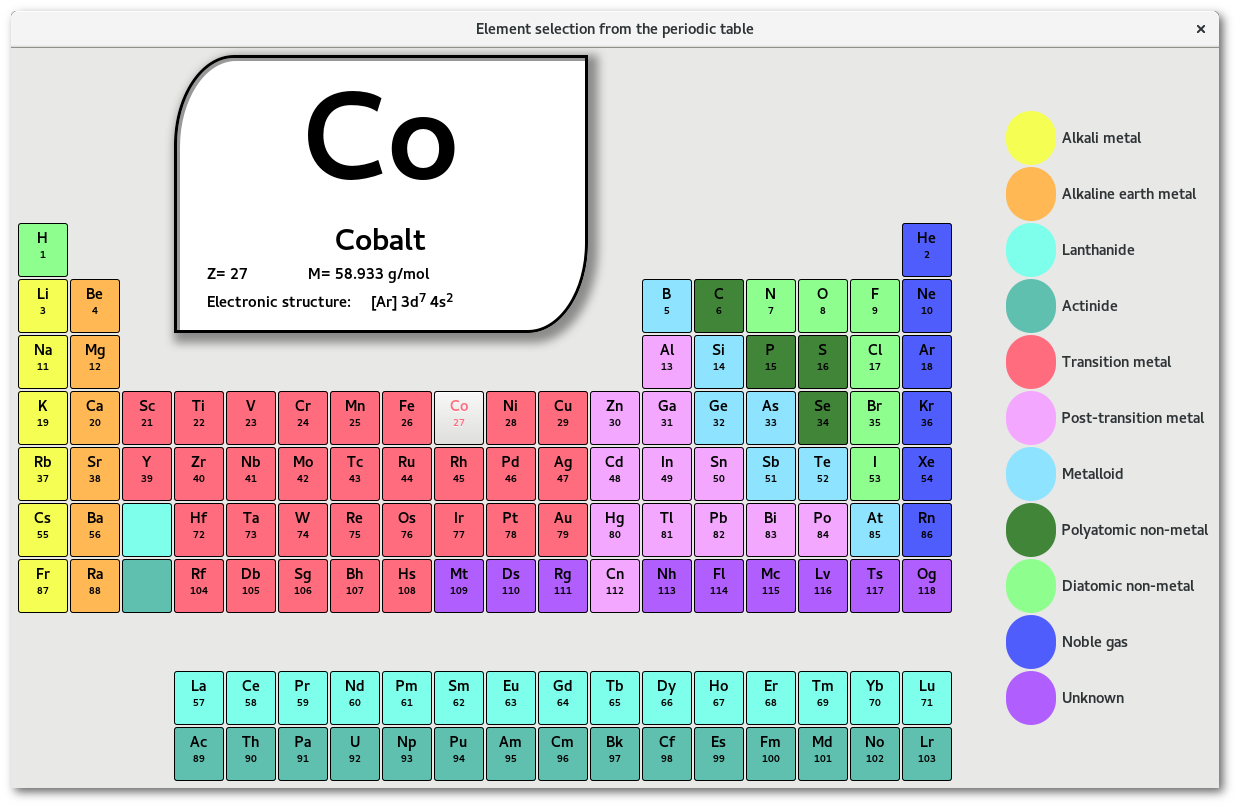
The "
Library" submenu [Fig. 6.17-d], to insert an new molecule in place of the object(s) to be removed. Some shortcuts are proposed as well, and the "More ..." button allows to open the "Library" dialog: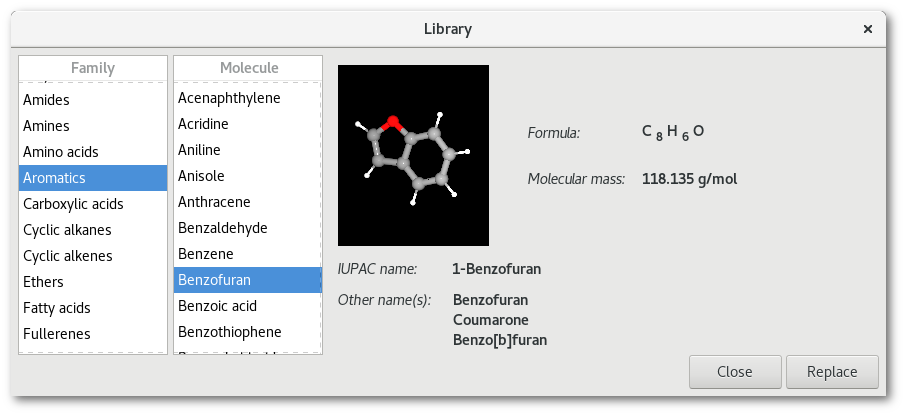
The "
Library" dialog provides a bunch of sample molecules roughly sorted by chemical properties (see appendix 3 for more details).The "
Import from project" submenu [Fig. 6.17-e], to insert atom(s) from any Atomes project opened in the workspace.The "
Copied data" button [Fig. 6.17-f], to copied data selection from any workspace in Atomes.
An example is provided in figure 6.18 with the random substitution of 500 "Si" atoms from the "SiO2" project by "water" molecules.
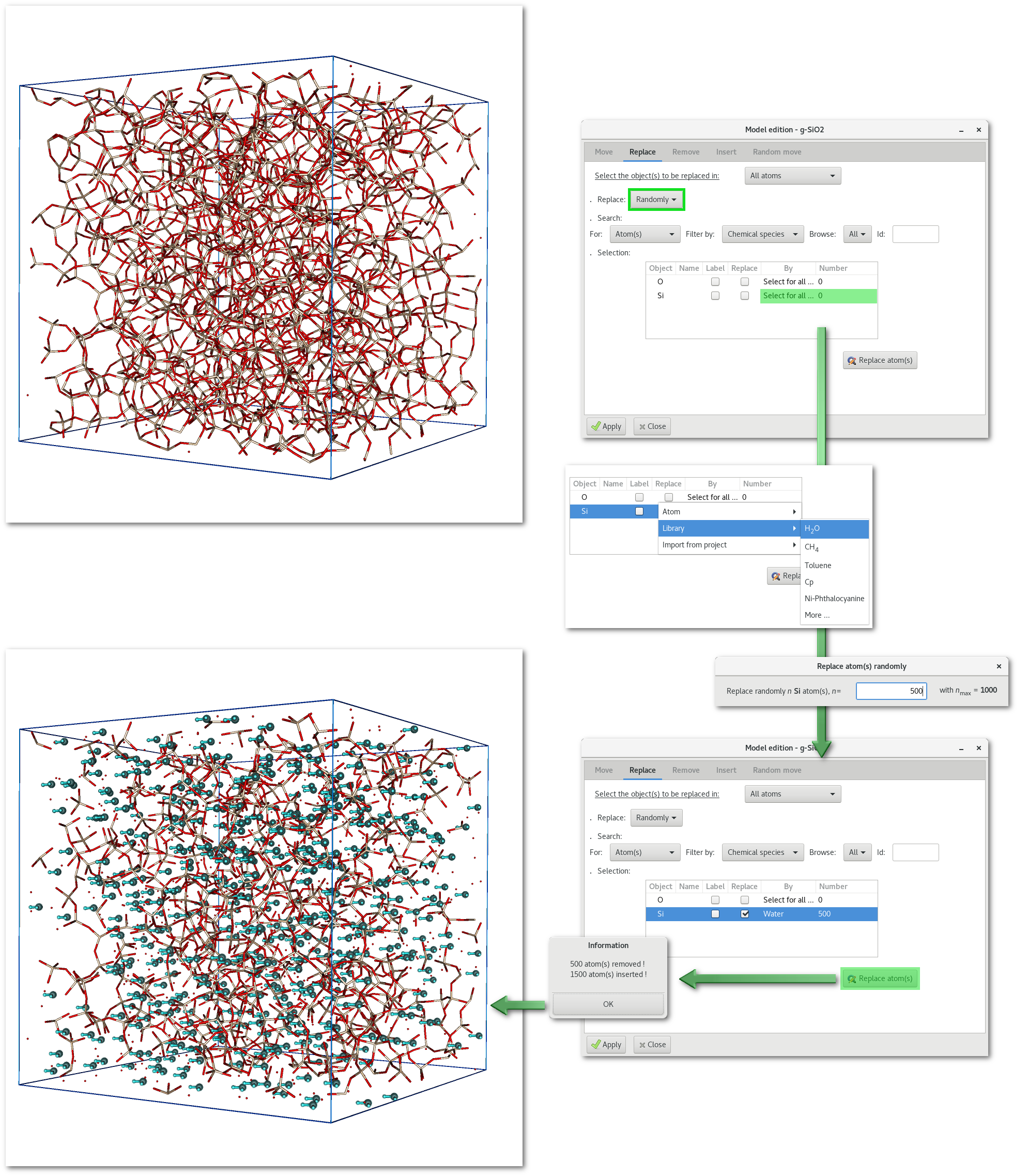
Irreversible: the replacement is irreversible (at least not directly, but it is always possible to perform another backward substitution) therefore remember to save your work before replacing any object(s) in your model.
Selection: At the end of the substitution process, and therefore after the insertion of the new objects, all these new objects will appear surrounded by light blue color in the OpenGL window (see figure 6.18), and will therefore be selected, making further work on these new objects easier.
Barycenters: the new object is inserted and centered at the barycenter of the atomic positions of the old object to be removed.
The "Remove" tab
The "Remove" tab allows to remove object(s), atom(s) or group of atoms, like for the "Replace" tab the action can also be performed randomly. An example is provided in figure 6.19 with the normal (one by one) removal of all "Ni-[N4]" coordination spheres atoms from the "Ni-Phth" project: for a Nickel atom, this means that this "Ni" atom as well as it surrounding "N" neighbours are all removed.
![Normal removal of all "Ni-[N_4]" coordination spheres from the model.](/Atomes-doc/img/edit/atoms/removex.png)
The "Insert" tab
The "Insert" tab allows to insert new object(s) in the model:
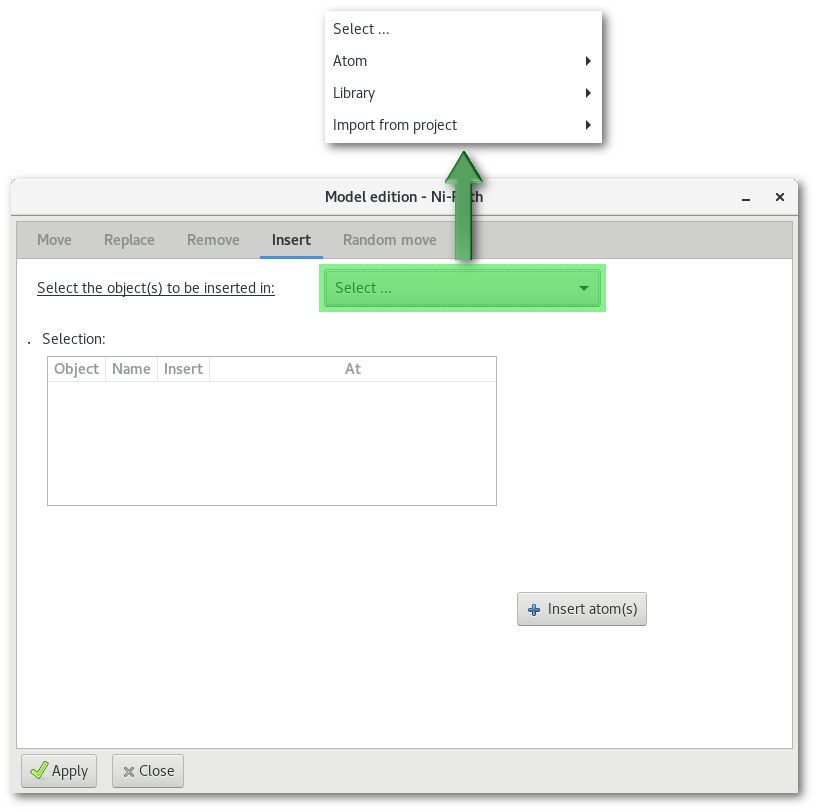
The "Select ..." menu reproduces the one from the "Replace" tab, see the bottom part in figure 6.17, and allows to insert atom(s) [Fig. 6.17-c], molecular fragment(s) [Fig. 6.17-d] and atom(s) from any other project opened in the workspace [Fig. 6.17-e]. Selecting any object to insert will populate and update the tree bellow, with a new line. Each line describes the object to be inserted, and offers options to confirm the insertion and specify the position where to insert the object in the model.
An example is provided in figure 6.21 where a fullerene "C240" is inserted in the "Ni-Phth" project modified after the removal example illustrated in figure 6.19.
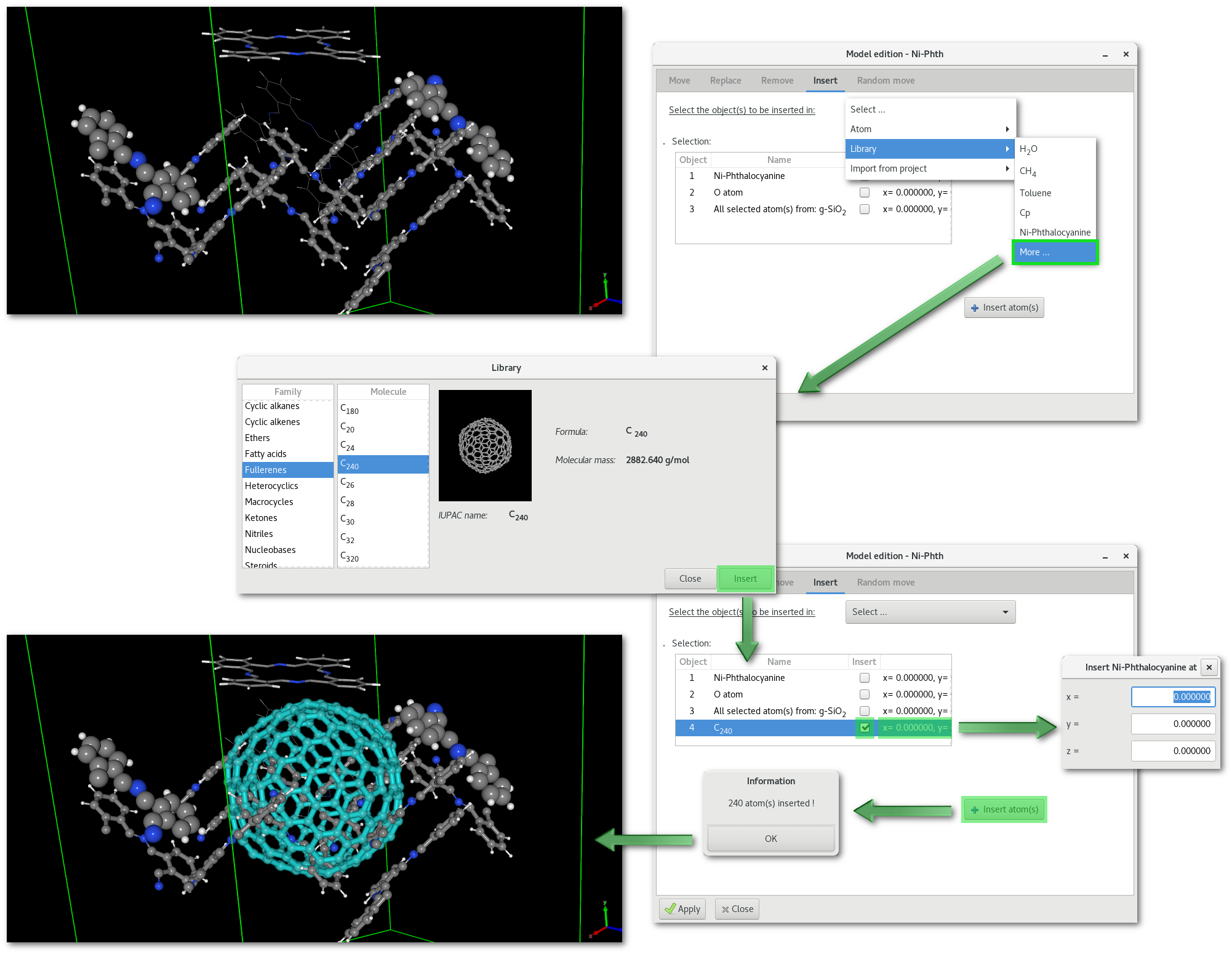
Irreversible: the insertion is irreversible (at least not directly, but it is always possible to remove all inserted atoms) therefore remember to save your work before inserting any object(s) in your model.
Selection: After the insertion of the new objects, all these new objects will appear surrounded by light blue color (see figure 6.21)in the OpenGL window, and will therefore be selected, making further work on these new objects easier.
Position: the coordinates (0.0, 0.0, 0.0) always refers to the center of the model, ie. the barycenter of all existing atomic coordinates.
The "Random move" tab
The "Random move" tab allows to disorder the model:
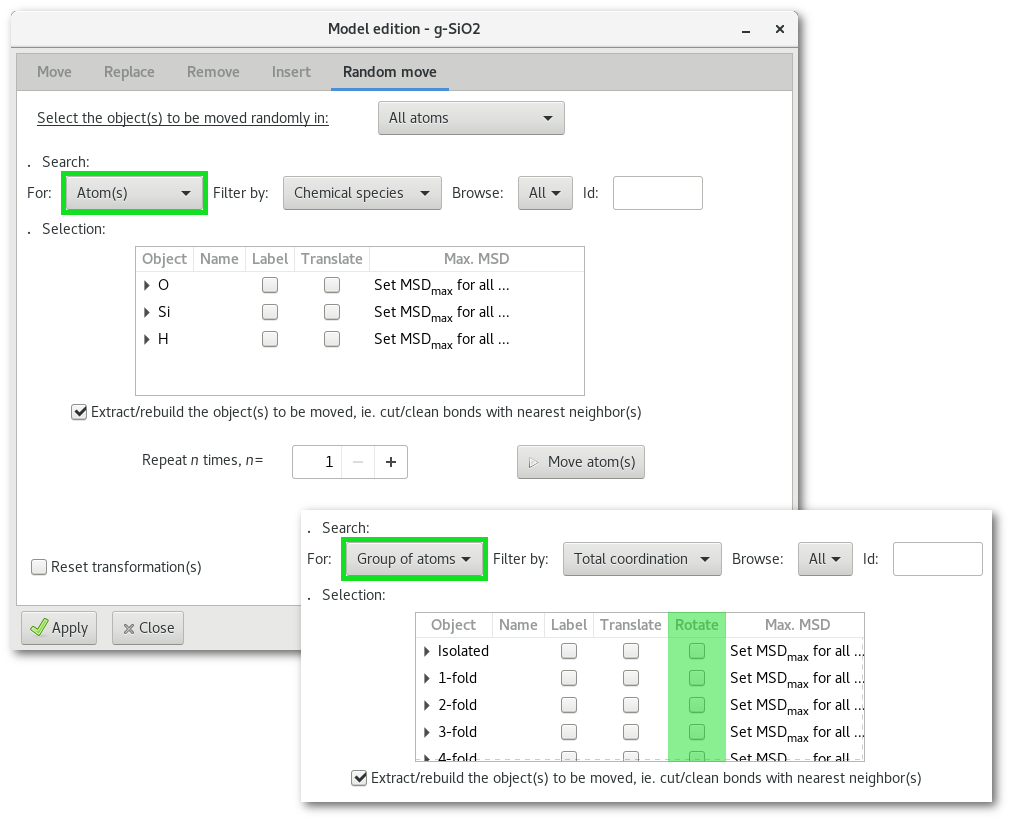
An example is provided in figure 6.23 with the random rotation of "water" molecules in the modified "SiO2" project after the substitution example illustrated in figure 6.18. Indeed after the substitution "water" molecules were all perfectly aligned, to correct this unlikely organization Atomes can rotate randomly and independently every water molecule in the model:
After the substitution all the "
H" and "O" atoms of the newly inserted "water" molecules are selected, so search for selected atom(s) only [Fig. 6.13-1].Then choose to apply the action to "
Group of atoms" [Fig. 6.13-2].Filter the selection using the "
Fragment" option [Fig. 6.13-3].After the steps [ran1], [ran2] and [ran3] the selection tree will correspond to the one in figure 6.23. To enable the random rotation simply click on the top header of the "
Rotate" column to select all object(s), actually all "water" molecules. It is required to enter a maximum MSD for each motion, click the top header of the "MAX. MSD" column to enter a suitable value. Optionally it is possible to iter the rotation process, at each step the rotation will be completely random.

Group of atoms: trying to move group of atoms that share atom(s) (ex: 2 coordination spheres with atom(s) in both coordination spheres) can lead to inaccurate results, the shared atom(s) being moved with each object they belong to.
Rotation: the rotation is performed around the positional barycenter of the object's atomic coordinates. In the case of a repeated rotation the rotation center is preserved until the final step to avoid drifting. If both translation and rotation are repeated then the object is translated first, updating as well the position of the rotation center, and then rotated.
Reconstruction: the check button labelled "
Extract/rebuild the object(s) to be moved, ie. cut/clean bonds with nearest neighbor(s)" offers the following option:For "
Atom(s)": if activated the object(s) to be moved will be extracted from the model and translated independently, otherwise chemical bond(s) will simply be stretched.For "
Group of atom(s)": if activated the atomic positions of the object(s) will be corrected to get only single piece object(s).