CPMD v4.3.0
The Atomes helper for the CPMD calculation offers to prepare input files for CPMD v4.3.0. Please note that the CPMD code offers so many calculation options that it is not possible either to provide a description or to offer a comprehensive usage guide for each of these options. Therefore the CPMD calculation assistant only provides help towards basics and / or frequently used calculation options.
In any case if you intent to use the CPMD code please refer to the user manual:
https://www.cpmd.org/wordpress/CPMD/getFile.php?file=manual.pdf
The Atomes helper for the CPMD calculation provides a step by step interface to configure the different sections of the CPMD input file:
The "INFO" section
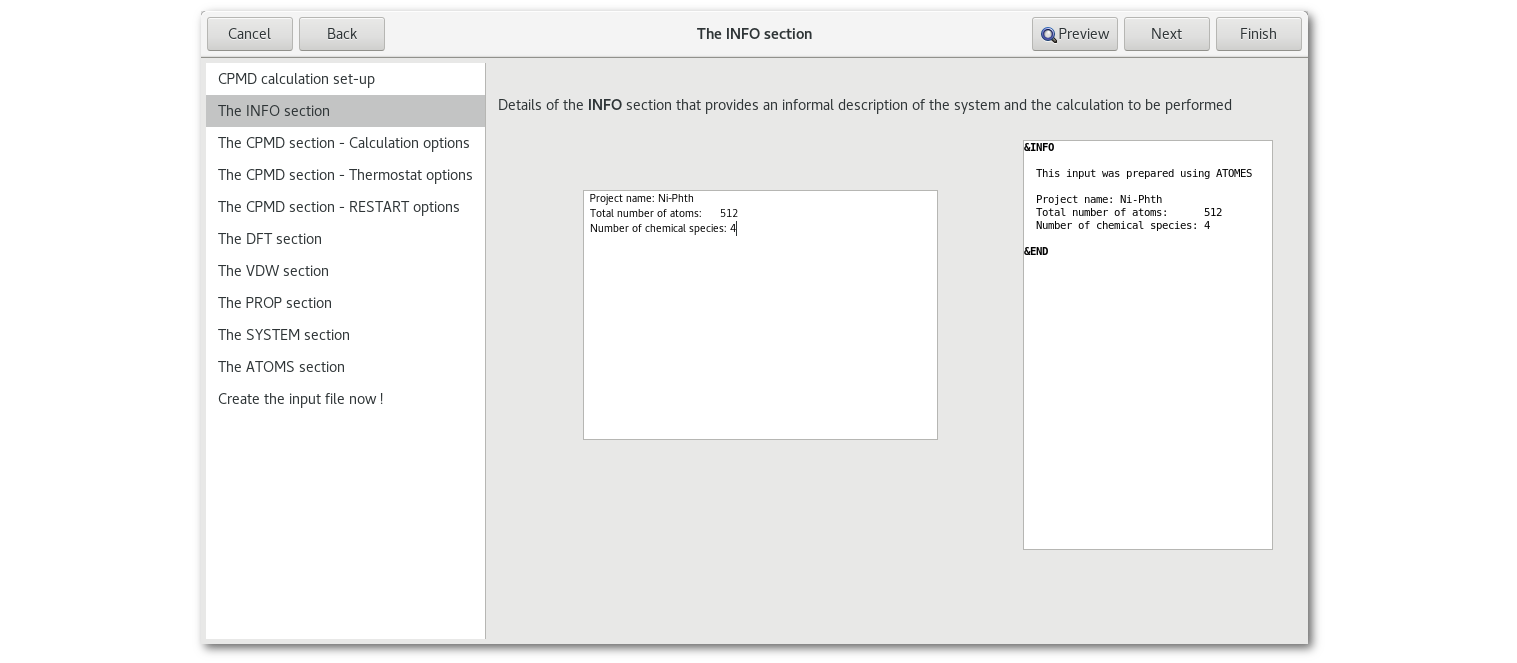
The "CPMD" section
The creation of the "CPMD" section is split over 3 tabs, dedicated respectively to the calculation, thermostat and restart option(s).
"Calculation options"
The "Calculation to be performed" [Fig. 7.36] combo allows to select the type of job:
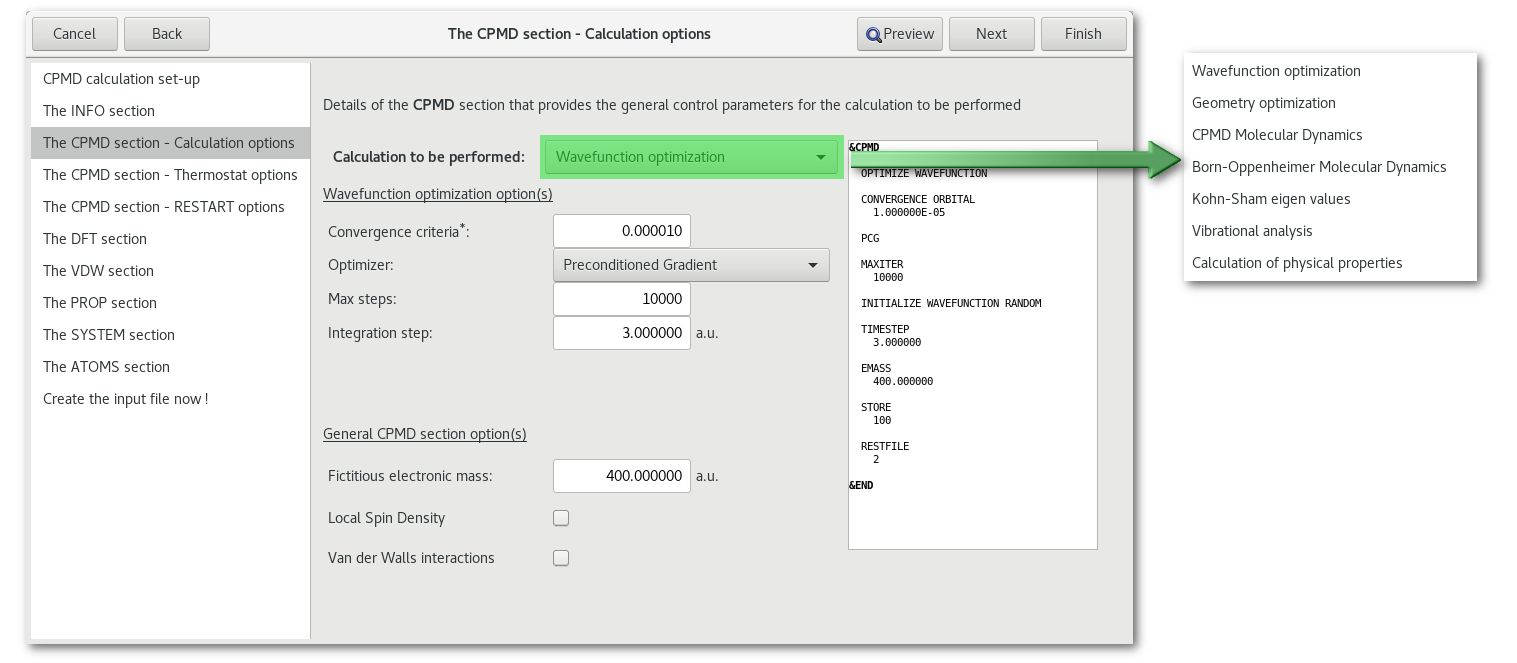
The option(s) directly bellow the combo will change depending on the nature of the calculation, please refer to the CPMD user manual for more information on theses options.
"Thermostat options"
If a molecular dynamics calculation is to be performed then the "CPMD section - Thermostat options" tab becomes accessible:
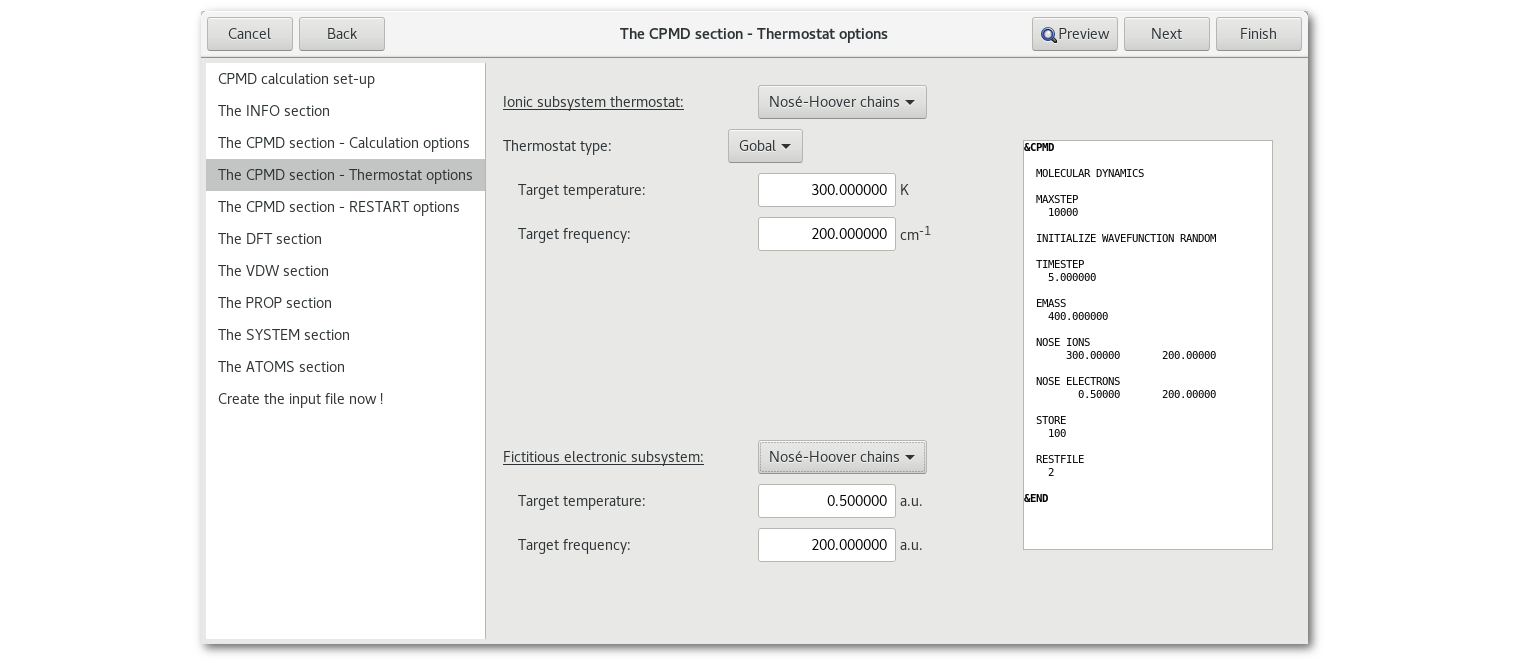
In order to access the next page of the assistant thermostat(s) must be set up properly. Again for more information please refer to the CPMD user manual.
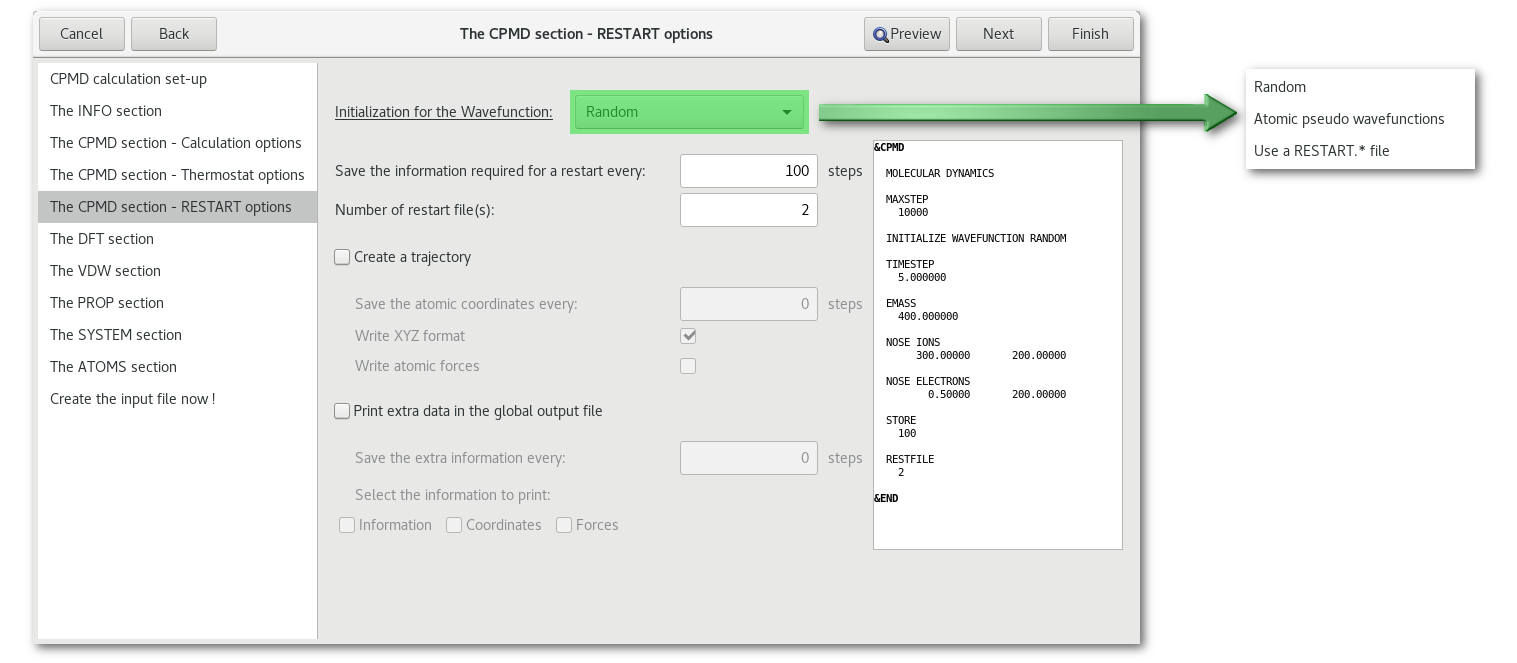
"RESTART options"
The "CPMD section - Restart options" tab present options to restart calculation and saving options during the calculation:
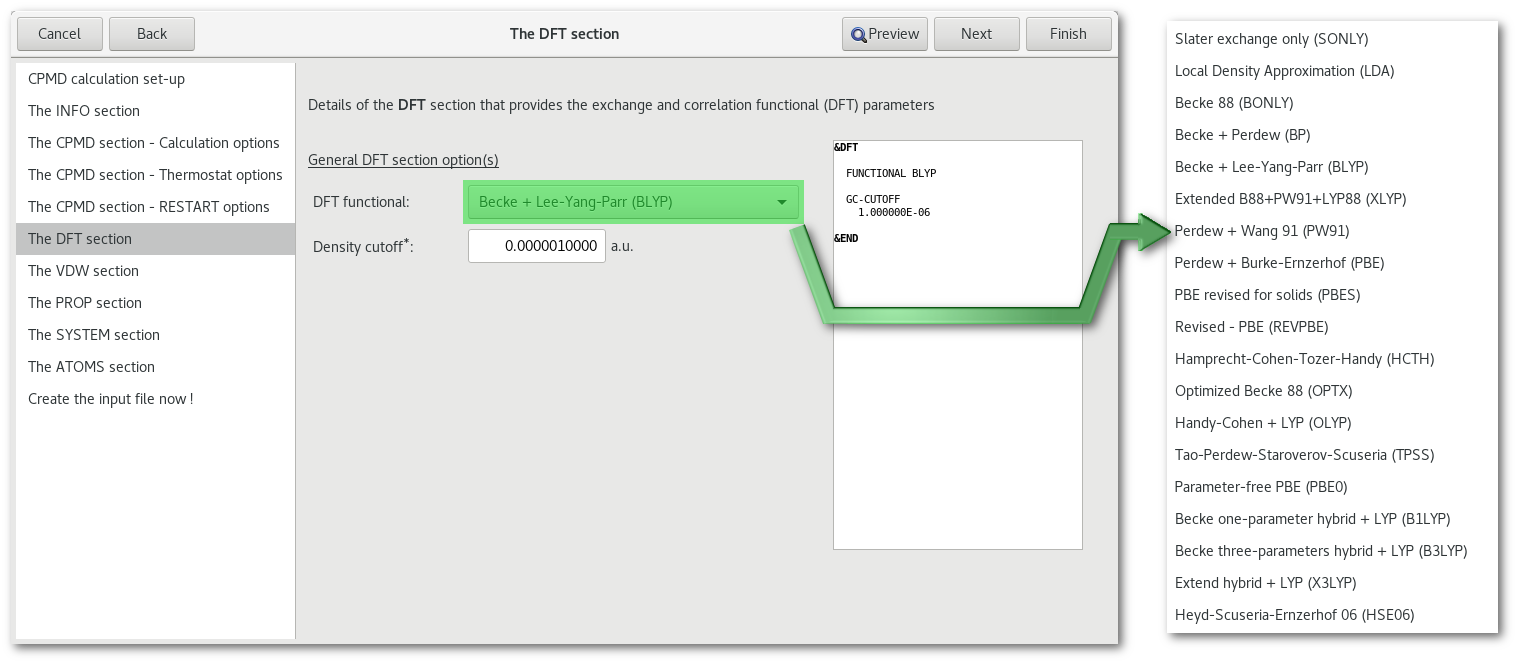
The "DFT" section
The "VDW" section
Providing that van der Waals interactions are selected in the "CPMD" section of the input file, then the "VDW" section becomes accessible. However its content is filled automatically based on parameters selected in both the "CPMD" and "DFT" sections.
The "PROP" section
Providing that the "Calculation of physical properties" is selected in the "CPMD" section of the input file, then the "PROP" section becomes accessible. However its content is filled automatically based on parameters selected in the "CPMD" section.
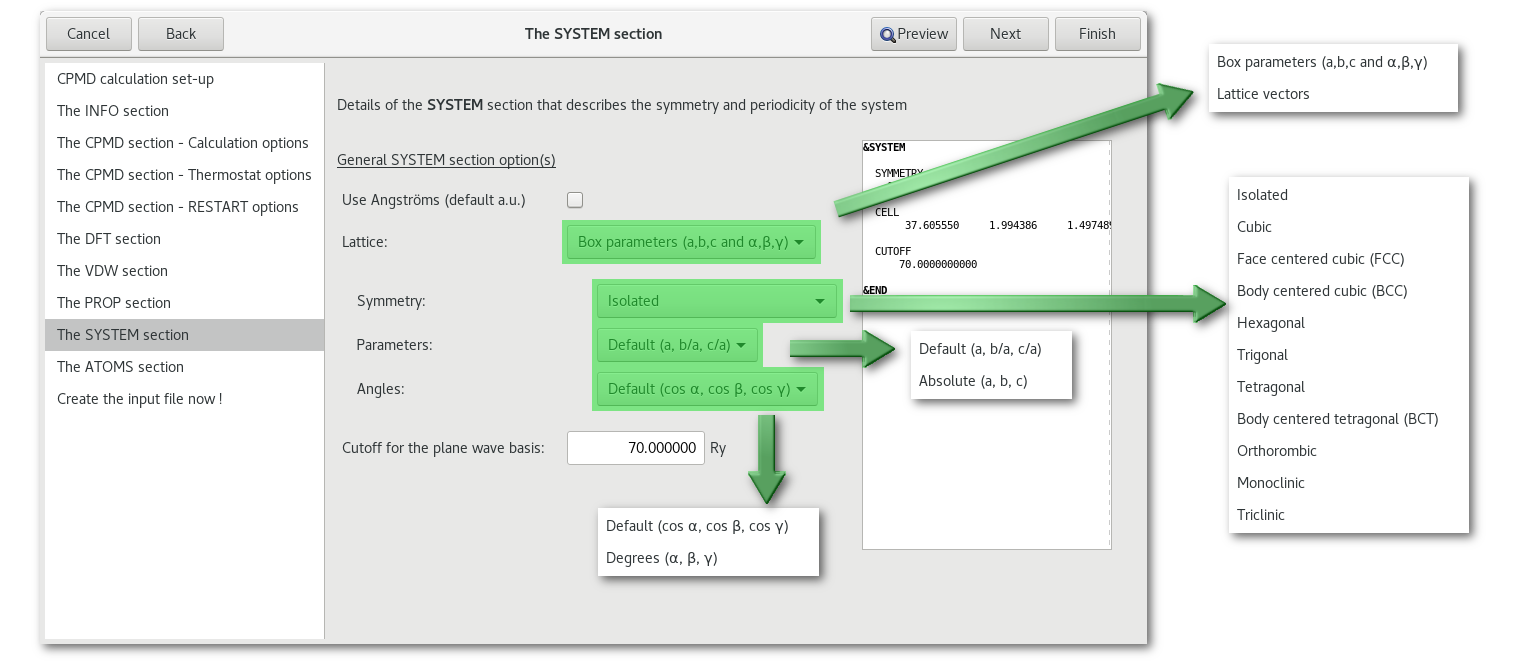
The "SYSTEM" section
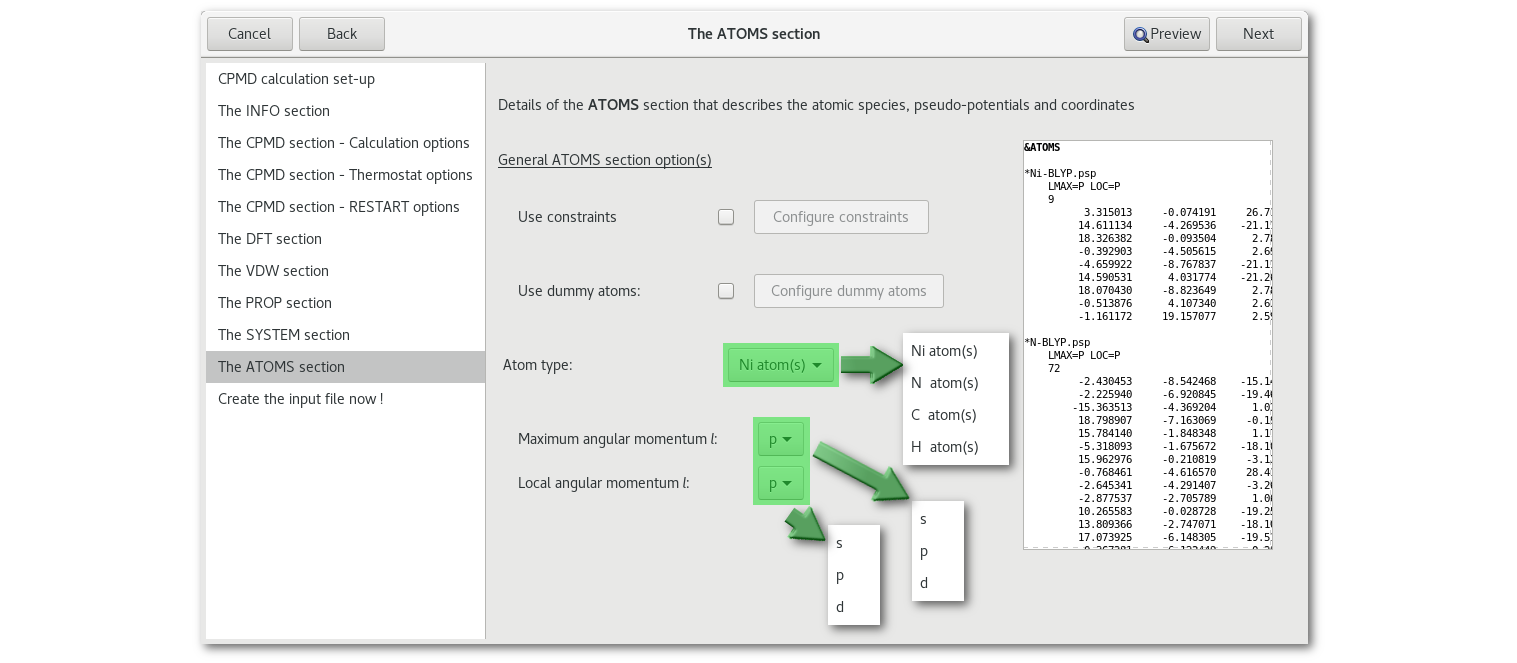
The "ATOMS" section